scATrans without spliced/unspliced layers: differential expression + enrichment + plotting#
This tutorial is for the majority of scRNA-seq users who do not have RNA
velocity (spliced/unspliced) layers — just an ordinary count matrix from
10x, Smart-seq, or any other protocol. Everything below uses
scat.differential_expression(...) instead of scat.active_score(...), and
never touches a spliced/unspliced/mature/nascent layer. The exact
same code works unmodified on a plain count-matrix .h5ad with no velocity
layers at all.
For the velocity-aware active_score workflow on the same dataset, see the
sibling tutorial Active Transcription Scoring on Real Spinal Cord Injury Data.
Dataset#
We use EC.h5ad: endothelial cells (EC) subset from a real mouse spinal
cord single-nucleus RNA-seq dataset, comparing uninjured controls (UN, 3
replicates) against spinal cord injury (SCI, 3 replicates):
Squair, J.W., Gautier, M., Kathe, C., et al. (2021). Confronting false discoveries in single-cell differential expression. Nature Communications 12, 5692. DOI: 10.1038/s41467-021-25960-2. GEO: GSE165003.
See References & Data Sources for the full citation list. This paper is itself about pseudoreplication and false-discovery risk in single-cell DE — fitting context for this tutorial, which reports real, sometimes non-significant results below rather than a cherry-picked toy example (small-n designs like this 3-vs-3 comparison commonly have limited genome-wide DE power; see Statistical Guidance & Reporting Checklist).
differential_expression and active_score share the same downstream
tooling — filter_active_genes, all scat.pl.* plots, and every enrichment
function — the only difference is that differential_expression skips the
unspliced-excess term entirely.
%matplotlib inline
import sys
sys.path.insert(0, "../../src") # use the in-repo scatrans, not any installed copy
import warnings
warnings.filterwarnings("ignore")
import scanpy as sc
import scatrans as scat
print("scatrans:", scat.__file__)
scatrans: /home/lieber/scATrans-main/docs/tutorials/../../src/scatrans/__init__.py
Load and inspect#
We deliberately only ever look at adata.X (raw counts) below — no
layers access anywhere in this notebook.
adata = sc.read_h5ad("../../EC.h5ad")
adata
AnnData object with n_obs × n_vars = 177 × 26451
obs: 'n_genes_by_counts', 'log1p_n_genes_by_counts', 'total_counts', 'log1p_total_counts', 'pct_counts_in_top_50_genes', 'pct_counts_in_top_100_genes', 'pct_counts_in_top_200_genes', 'pct_counts_in_top_500_genes', 'condition', 'sample', 'GSM_ID', 'total_counts_mt', 'pct_counts_mt', 'n_genes', 'doublet_score', 'predicted_doublet'
var: 'gene_symbol', 'mt', 'n_cells_by_counts', 'mean_counts', 'pct_dropout_by_counts', 'total_counts', 'n_cells'
uns: 'sample_colors', 'scrublet'
layers: 'ambiguous', 'mature', 'nascent', 'spliced', 'unspliced'
adata.obs[["condition", "sample", "GSM_ID"]].value_counts().sort_index()
condition sample GSM_ID
SCI rep1 GSM5024314 35
rep2 GSM5024315 23
rep3 GSM5024316 37
UN rep1 GSM5024317 27
rep2 GSM5024318 38
rep3 GSM5024319 17
Name: count, dtype: int64
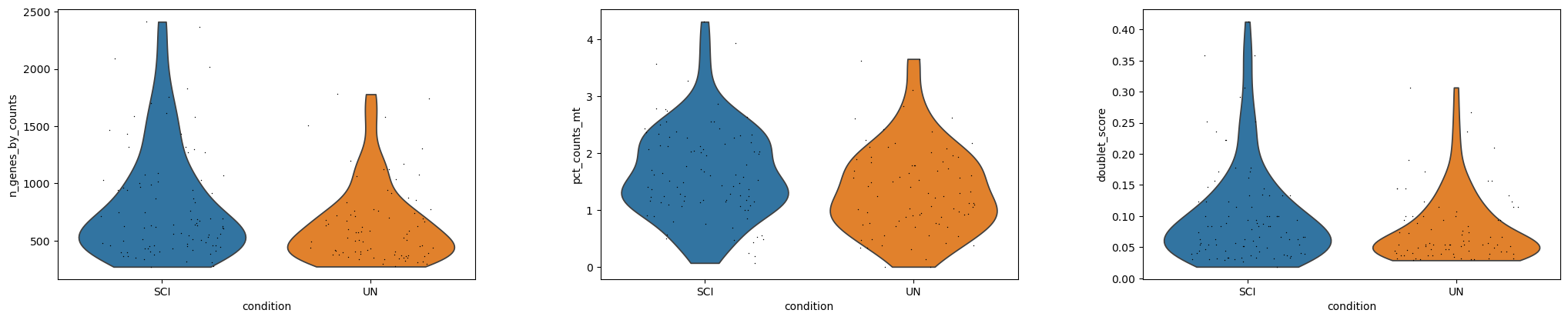
QC and light filtering#
This EC subset was already QC’d upstream (doublet scores, mito %, and gene
counts are precomputed in .obs). We apply the same filters here for
completeness / robustness — on this dataset they happen to remove zero
cells, since the upstream authors already excluded low-quality cells.
n0 = adata.n_obs
adata = adata[~adata.obs["predicted_doublet"]].copy()
adata = adata[adata.obs["pct_counts_mt"] < 20].copy()
adata = adata[adata.obs["n_genes_by_counts"] > 200].copy()
sc.pp.filter_genes(adata, min_cells=3)
print(f"cells kept: {adata.n_obs} of {n0}")
print(f"genes kept: {adata.n_vars}")
cells kept: 177 of 177
genes kept: 9221
sc.pl.violin(
adata, ["n_genes_by_counts", "pct_counts_mt", "doublet_score"],
groupby="condition", jitter=0.3, multi_panel=True,
)
Preserve raw counts, then preprocess#
store_raw_counts snapshots .X into layers["counts"] (and .raw) before
any HVG/normalize/log1p step — this is what lets enrichment functions later
auto-supply the correct measured-gene background via adata=. Count-based
DE backends (PyDESeq2, Memento) need these raw integer counts; the
de_preprocess="auto" step inside differential_expression also uses the
.uns["log1p"] marker scanpy sets to detect whether .X is already
log-normalized (see Standalone Differential Expression (no velocity data required) for the
anndata.concat() caveat around this marker).
scat.store_raw_counts(adata, layer="counts", save_raw=True)
adata_norm = adata.copy()
sc.pp.normalize_total(adata_norm, target_sum=1e4)
sc.pp.log1p(adata_norm)
Backend A: single-cell Wilcoxon DE#
The simplest, fastest path: scanpy’s rank_genes_groups directly on
log-normalized single cells.
adata_norm, de_wilcoxon = scat.differential_expression(
adata_norm,
groupby="condition",
target_group="SCI",
reference_group="UN",
de_method="wilcoxon",
de_preprocess="none", # we already normalized + log1p'd above
)
de_wilcoxon.sort_values("logFC", ascending=False).head(10)
WARNING: It seems you use rank_genes_groups on the raw count data. Please logarithmize your data before calling rank_genes_groups.
| logFC | p_val | p_adj | baseMean | |
|---|---|---|---|---|
| Tpcn1 | 27.242462 | 0.227766 | 0.9938 | 0.116235 |
| Prdm16 | 27.127773 | 0.334591 | 0.9938 | 0.118895 |
| Pgls | 27.004536 | 0.227766 | 0.9938 | 0.126359 |
| Nxn | 27.004536 | 0.227766 | 0.9938 | 0.137124 |
| Gm57952 | 27.004536 | 0.227766 | 0.9938 | 0.124299 |
| Rab11fip1 | 27.004536 | 0.277691 | 0.9938 | 0.103826 |
| Dmtf1 | 27.004536 | 0.147796 | 0.9938 | 0.125101 |
| Pacsin2 | 26.871260 | 0.334591 | 0.9938 | 0.104579 |
| Kcnj10 | 26.871260 | 0.227766 | 0.9938 | 0.122506 |
| Plcl1 | 26.871260 | 0.277691 | 0.9938 | 0.112288 |
print("min p_adj:", de_wilcoxon["p_adj"].min())
print("genes with p_adj < 0.05:", (de_wilcoxon["p_adj"] < 0.05).sum())
min p_adj: 0.9938003852887332
genes with p_adj < 0.05: 0
Note
With only 3 replicates per condition, no gene reaches genome-wide
p_adj < 0.05 here — this is real, honest small-n biology, not a bug.
filter_active_genes with strict cutoffs legitimately returns very few or
zero candidates. The practical workaround (used throughout this notebook)
is to rank by nominal (unadjusted) p-value and effect size for
hypothesis-generating candidate lists, and to say so explicitly rather
than reporting p_adj < 0.05 claims that the data does not support. See
Statistical Guidance & Reporting Checklist for the full reporting checklist.
candidates_strict = scat.filter_active_genes(de_wilcoxon, pval_cutoff=0.2, logfc_cutoff=0.3)
print("strict candidates (p_adj<0.2, |logFC|>0.3):", len(candidates_strict))
# Exploratory ranking: top 100 upregulated genes by nominal p-value
candidates = de_wilcoxon[de_wilcoxon["logFC"] > 0].sort_values("p_val").head(100)
print("exploratory candidates (top 100 upregulated by nominal p-value):", len(candidates))
candidates.head(10)
strict candidates (p_adj<0.2, |logFC|>0.3): 0
exploratory candidates (top 100 upregulated by nominal p-value): 100
| logFC | p_val | p_adj | baseMean | |
|---|---|---|---|---|
| Rpl37 | 2.161662 | 0.000302 | 0.9938 | 1.468993 |
| Spock2 | 2.577669 | 0.000312 | 0.9938 | 1.783297 |
| Rpl35 | 2.087135 | 0.000397 | 0.9938 | 1.085354 |
| Igf1r | 1.444869 | 0.000446 | 0.9938 | 1.513025 |
| Gpcpd1 | 1.772408 | 0.000603 | 0.9938 | 1.247660 |
| Rpl37a | 2.572021 | 0.001323 | 0.9938 | 1.647515 |
| Rpl38 | 2.344653 | 0.001573 | 0.9938 | 1.909690 |
| Gm68844 | 2.785950 | 0.001696 | 0.9938 | 1.732615 |
| Rps19 | 1.891919 | 0.002009 | 0.9938 | 1.650908 |
| Cpe | 2.122542 | 0.002111 | 0.9938 | 0.719209 |
Backend B: pseudobulk + PyDESeq2#
With 3 biological replicates per group, aggregating to pseudobulk and running count-based DESeq2 is the more defensible choice for a real publication (see the DE-backend decision guide in Core Workflow). We compare its top genes against the Wilcoxon single-cell result above — backend choice visibly changes which genes come out on top.
adata_pb = adata.copy()
adata_pb, de_pseudobulk = scat.differential_expression(
adata_pb,
groupby="condition",
target_group="SCI",
reference_group="UN",
use_pseudobulk=True,
sample_col="sample",
pseudobulk_de_backend="pydeseq2",
)
print("min p_adj (pseudobulk):", de_pseudobulk["p_adj"].min())
de_pseudobulk.sort_values("logFC", ascending=False).head(10)
min p_adj (pseudobulk): 0.5584481513291821
| logFC | p_val | p_adj | baseMean | |
|---|---|---|---|---|
| Prdm16 | 4.416334 | 0.043152 | 0.999513 | 2.833333 |
| Jmy | 4.315898 | 0.050361 | 0.999513 | 3.000000 |
| Gm3764 | 4.147823 | 0.062356 | 0.999513 | 2.666667 |
| Col4a3 | 3.979546 | 0.080037 | 0.999513 | 2.500000 |
| Cd38 | 3.958031 | 0.080950 | 0.999513 | 2.000000 |
| Fau | 3.825012 | 0.093960 | 0.999513 | 2.000000 |
| Gm57952 | 3.819496 | 0.096126 | 0.999513 | 2.000000 |
| Itih3 | 3.744748 | 0.113096 | 0.999513 | 1.833333 |
| Gkn3 | 3.726783 | 0.114265 | 0.999513 | 2.166667 |
| Msantd4 | 3.667388 | 0.114968 | 0.999513 | 1.666667 |
top_wilcoxon = set(de_wilcoxon.sort_values("logFC", ascending=False).head(20).index)
top_pseudobulk = set(de_pseudobulk.sort_values("logFC", ascending=False).head(20).index)
print(f"overlap of top-20 genes (Wilcoxon vs. pseudobulk PyDESeq2): {len(top_wilcoxon & top_pseudobulk)} / 20")
overlap of top-20 genes (Wilcoxon vs. pseudobulk PyDESeq2): 7 / 20
Backend C: Memento (method-of-moments cell-level DE)#
Memento (Cell, 2024) is a
method-of-moments estimator that works directly on raw single-cell counts
(no pseudobulk aggregation, no log-normalization). It needs use_memento_de=True
and raw integer counts, which is exactly why we called store_raw_counts
above.
adata_mem = adata.copy()
adata_mem, de_memento = scat.differential_expression(
adata_mem,
groupby="condition",
target_group="SCI",
reference_group="UN",
use_memento_de=True,
)
de_memento.sort_values("logFC", ascending=False).head(10)
[Parallel(n_jobs=-1)]: Using backend LokyBackend with 12 concurrent workers.
[Parallel(n_jobs=-1)]: Done 26 tasks | elapsed: 1.4s
[Parallel(n_jobs=-1)]: Done 476 tasks | elapsed: 2.9s
[Parallel(n_jobs=-1)]: Done 1476 tasks | elapsed: 5.7s
[Parallel(n_jobs=-1)]: Done 2083 out of 2083 | elapsed: 7.5s finished
| logFC | p_val | p_adj | memento_de_se | memento_dv_coef | memento_dv_se | memento_dv_pval | baseMean | |
|---|---|---|---|---|---|---|---|---|
| Gfap | 2.883938 | 0.065733 | 0.987457 | 0.899865 | 1.124937 | 2.501670 | 0.873866 | 0.401130 |
| Slc6a20a | 2.396857 | 0.011514 | 0.666649 | 0.602903 | 1.624803 | 1.066352 | 0.204310 | 0.225989 |
| Lama4 | 2.044974 | 0.030588 | 0.810178 | 0.590835 | 0.891034 | 1.457454 | 0.750794 | 0.175141 |
| Mecom | 1.958642 | 0.008734 | 0.606432 | 0.492907 | 1.074449 | 1.579175 | 0.649023 | 0.288136 |
| Ptp4a2 | 1.812726 | 0.025814 | 0.810178 | 0.518275 | 1.446629 | 1.680044 | 0.983735 | 0.197740 |
| Mcf2l | 1.801148 | 0.086967 | 0.989809 | 0.644036 | -0.380125 | 1.863114 | 0.596109 | 0.276836 |
| Phf20l1 | 1.773532 | 0.224842 | 0.989809 | 0.801671 | -2.442645 | 1.840142 | 0.092357 | 0.158192 |
| Sar1b | 1.726292 | 0.026552 | 0.810178 | 0.518877 | 1.886126 | 1.580312 | 0.884016 | 0.152542 |
| Ifnar1 | 1.685207 | 0.035636 | 0.839459 | 0.518997 | 1.173949 | 1.289252 | 0.419856 | 0.225989 |
| Taok1 | 1.676860 | 0.136702 | 0.989809 | 0.678254 | -0.386383 | 2.072747 | 0.504406 | 0.163842 |
Three backends, three slightly different answers — this is exactly the kind of pseudoreplication-sensitivity that the Squair et al. (2021) paper behind this dataset warns about. Always report which backend was used, per Statistical Guidance & Reporting Checklist.
Functional enrichment: the full tour#
We use the 100-gene exploratory candidate list from Backend A for the ORA
methods below (run_enrichment, run_kegg, run_go), and the full ranked
logFC list for run_gsea.
candidate_genes = candidates.index.tolist()
down_genes = de_wilcoxon[de_wilcoxon["logFC"] < 0].sort_values("p_val").head(100).index.tolist()
len(candidate_genes), len(down_genes)
(100, 100)
Over-representation analysis (ORA): GO Biological Process#
go_res = scat.run_enrichment(
candidate_genes,
gene_sets="GO_Biological_Process",
organism="mouse",
adata=adata_norm, # auto-supplies the stored measured-gene background
return_all=True,
)
print(f"{len(go_res)} GO BP terms tested, {(go_res['p.adjust'] < 0.05).sum()} significant at p.adjust<0.05")
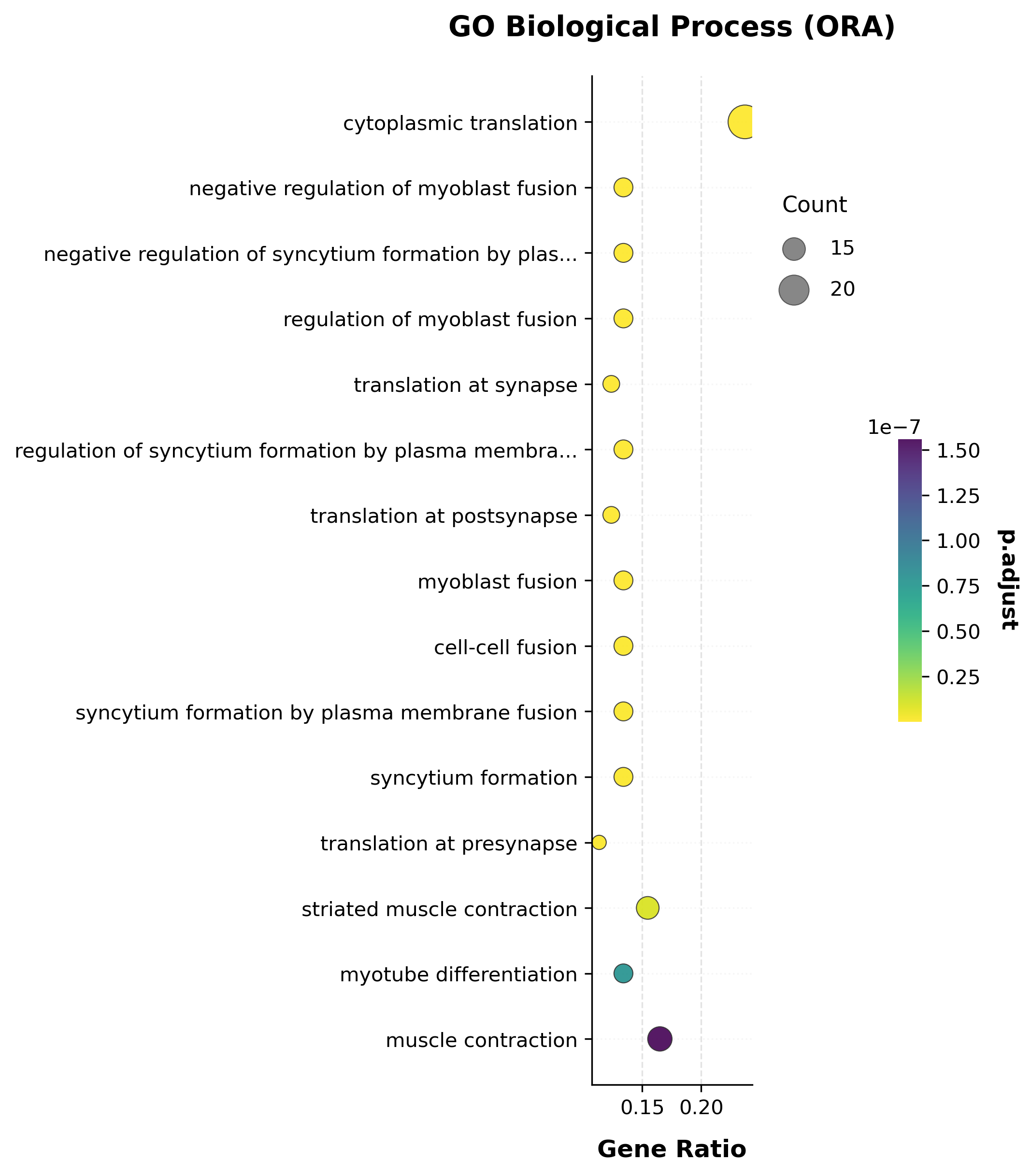
go_res.sort_values("pvalue").head(10)[["Term", "Count", "GeneRatio", "pvalue", "p.adjust"]]
6744 GO BP terms tested, 47 significant at p.adjust<0.05
| Term | Count | GeneRatio | pvalue | p.adjust | |
|---|---|---|---|---|---|
| 0 | cytoplasmic translation (GO:0002181) | 23 | 0.237113 | 3.700163e-17 | 2.495390e-13 |
| 1 | negative regulation of myoblast fusion (GO:190... | 13 | 0.134021 | 1.380523e-15 | 4.655122e-12 |
| 2 | negative regulation of syncytium formation by ... | 13 | 0.134021 | 2.655028e-15 | 5.968503e-12 |
| 3 | regulation of myoblast fusion (GO:1901739) | 13 | 0.134021 | 1.184686e-14 | 1.997380e-11 |
| 4 | translation at synapse (GO:0140241) | 12 | 0.123711 | 7.259096e-14 | 7.073882e-11 |
| 6 | translation at postsynapse (GO:0140242) | 12 | 0.123711 | 7.259096e-14 | 7.073882e-11 |
| 5 | regulation of syncytium formation by plasma me... | 13 | 0.134021 | 7.342405e-14 | 7.073882e-11 |
| 7 | myoblast fusion (GO:0007520) | 13 | 0.134021 | 1.860772e-13 | 1.568630e-10 |
| 8 | cell-cell fusion (GO:0140253) | 13 | 0.134021 | 1.443489e-12 | 9.734891e-10 |
| 9 | syncytium formation by plasma membrane fusion ... | 13 | 0.134021 | 1.443489e-12 | 9.734891e-10 |
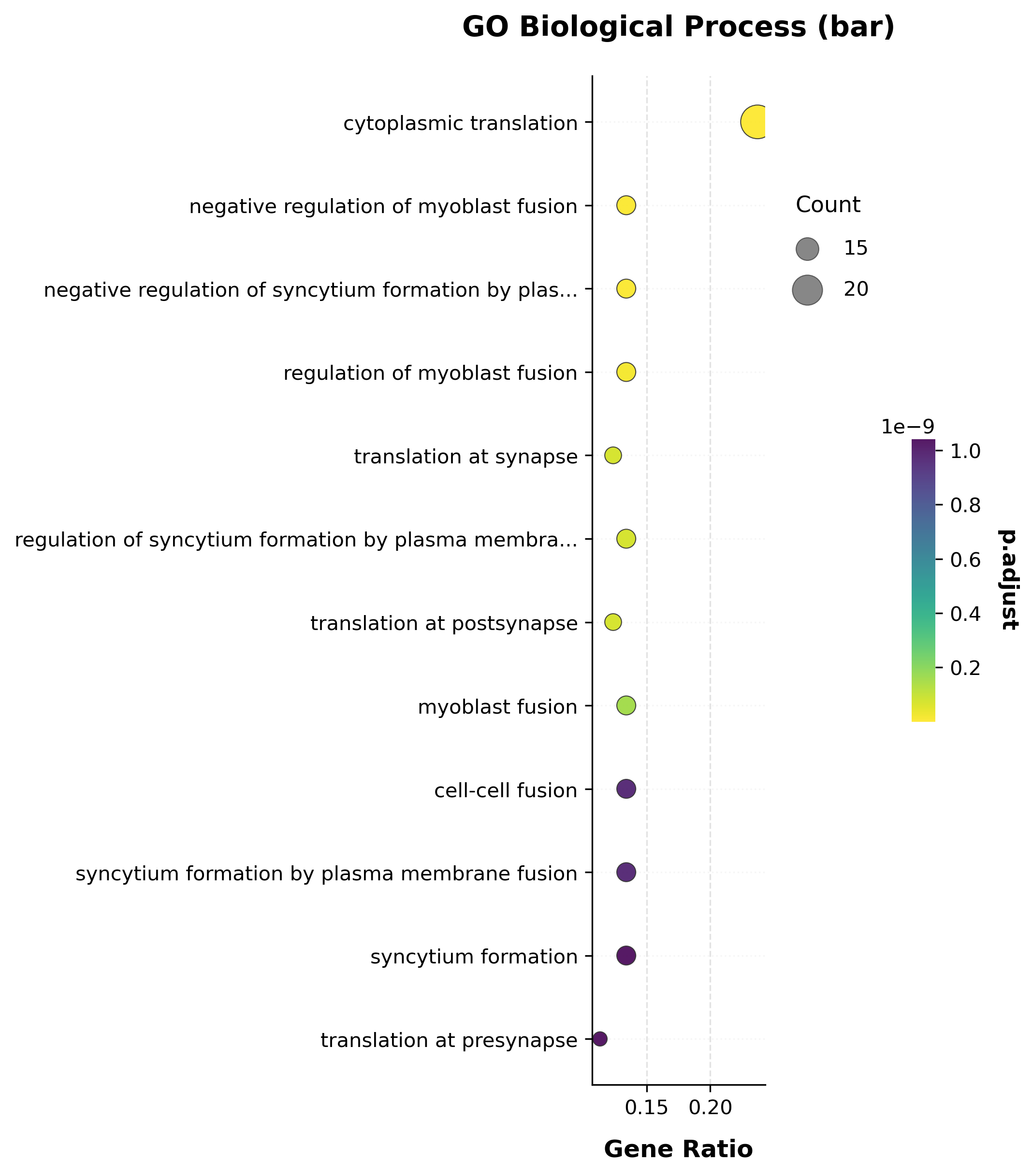
Several ribosomal-protein genes (Rpl37, Rpl35, Rpl37a, Rpl38,
Rps19, …) are in the candidate list, so “cytoplasmic translation” comes
out as the top term — unlike the per-gene DE p-values, gene-set-level ORA
here has genuine, well-powered signal.
KEGG pathways#
kegg_res = scat.run_kegg(
candidate_genes,
organism="mouse",
adata=adata_norm,
return_all=True,
)
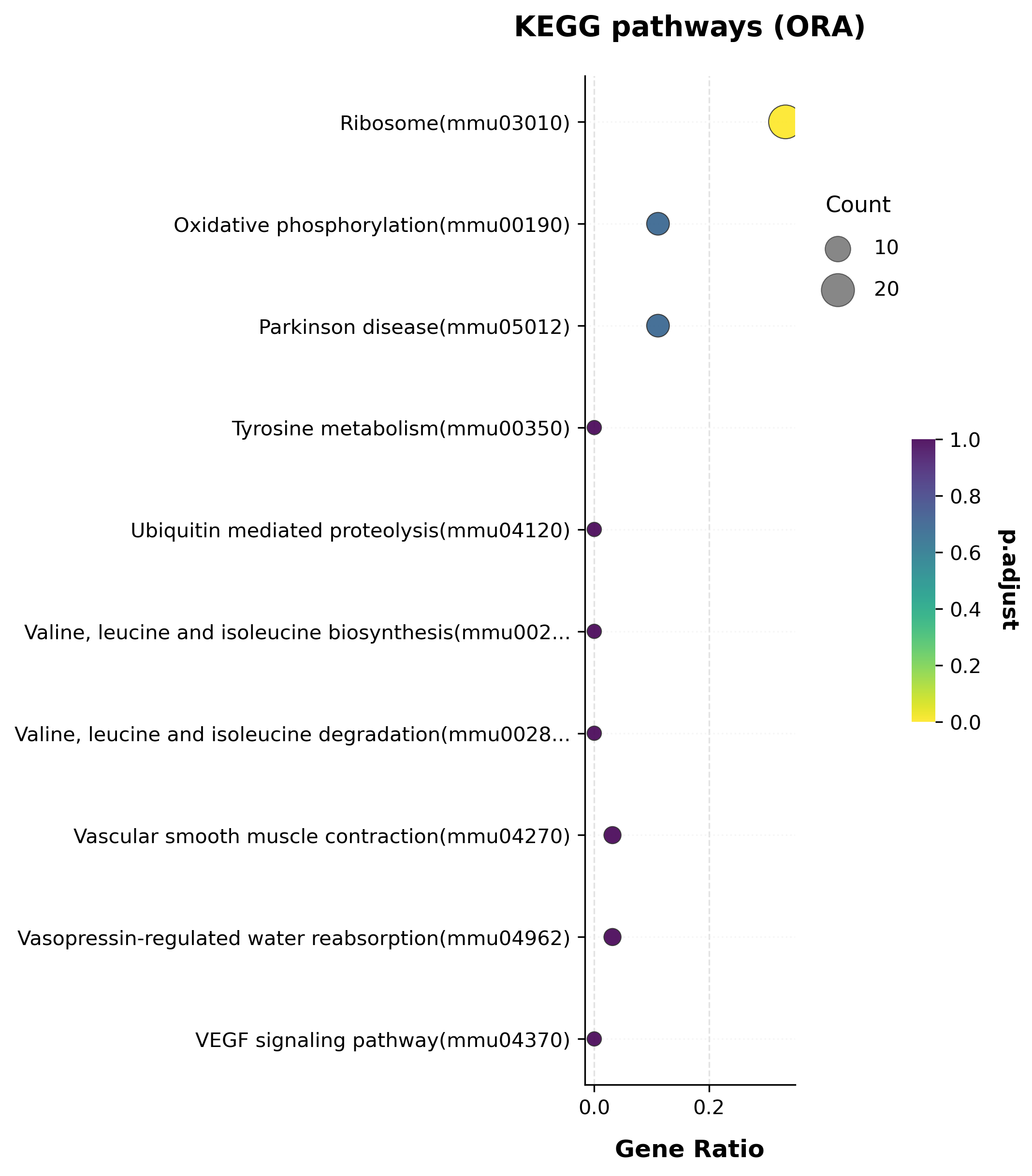
kegg_res.sort_values("pvalue").head(10)[["Term", "Count", "pvalue", "p.adjust"]]
| Term | Count | pvalue | p.adjust | |
|---|---|---|---|---|
| 0 | Ribosome(mmu03010) | 21 | 4.654655e-18 | 9.402402e-16 |
| 2 | Parkinson disease(mmu05012) | 7 | 9.762118e-03 | 6.892085e-01 |
| 1 | Oxidative phosphorylation(mmu00190) | 7 | 1.023577e-02 | 6.892085e-01 |
| 193 | Alzheimer disease(mmu05010) | 7 | 2.363543e-02 | 1.000000e+00 |
| 32 | Bile secretion(mmu04976) | 3 | 2.908569e-02 | 1.000000e+00 |
| 201 | ABC transporters(mmu02010) | 2 | 5.766020e-02 | 1.000000e+00 |
| 66 | Focal adhesion(mmu04510) | 6 | 6.453936e-02 | 1.000000e+00 |
| 38 | Cardiac muscle contraction(mmu04260) | 3 | 7.023010e-02 | 1.000000e+00 |
| 72 | Glioma(mmu05214) | 3 | 7.808674e-02 | 1.000000e+00 |
| 169 | Small cell lung cancer(mmu05222) | 3 | 9.056147e-02 | 1.000000e+00 |
“Ribosome” is overwhelmingly the top KEGG hit here (p.adjust ≈ 1e-15) —
consistent with the GO result above.
GO, all three ontologies at once (ontology="ALL")#
go_all = scat.run_go(
candidate_genes,
ontology="ALL",
organism="mouse",
adata=adata_norm,
return_all=True,
adjust_across_all=True, # single BH correction across BP+CC+MF combined
)
print("ontologies covered:", list(go_all.attrs["per_ontology_attrs"].keys()))
go_all.sort_values("pvalue").head(6)[["Term", "Count", "pvalue", "p.adjust"]]
ontologies covered: ['BP', 'CC', 'MF']
| Term | Count | pvalue | p.adjust | |
|---|---|---|---|---|
| 0 | cytoplasmic translation (GO:0002181) | 23 | 3.700163e-17 | 2.495390e-13 |
| 1 | negative regulation of myoblast fusion (GO:190... | 13 | 1.380523e-15 | 4.655122e-12 |
| 2 | negative regulation of syncytium formation by ... | 13 | 2.655028e-15 | 5.968503e-12 |
| 3 | regulation of myoblast fusion (GO:1901739) | 13 | 1.184686e-14 | 1.997380e-11 |
| 6 | translation at postsynapse (GO:0140242) | 12 | 7.259096e-14 | 7.073882e-11 |
| 5 | translation at synapse (GO:0140241) | 12 | 7.259096e-14 | 7.073882e-11 |
Pre-ranked GSEA#
run_gsea takes a full ranked gene list (here, logFC from the Wilcoxon
DE result) rather than a fixed candidate cutoff — every gene contributes,
weighted by its rank.
Note
nperm controls the permutation-based null distribution; we use a modest
nperm=100 here to keep this tutorial cell fast (real run: ~27s). For a
manuscript, prefer a larger nperm (≥1000). Roughly 88% of genes in this
ranked list share tied logFC values (a side effect of the small-n design
above), which run_gsea warns about — tie-breaking within those ranks is
arbitrary, so treat exact NES values here as illustrative rather than
final.
ranked = de_wilcoxon["logFC"].sort_values(ascending=False)
gsea_res = scat.run_gsea(
ranked_genes=ranked,
gene_sets="GO_Biological_Process",
organism="mouse",
nperm=100,
min_size=15,
)
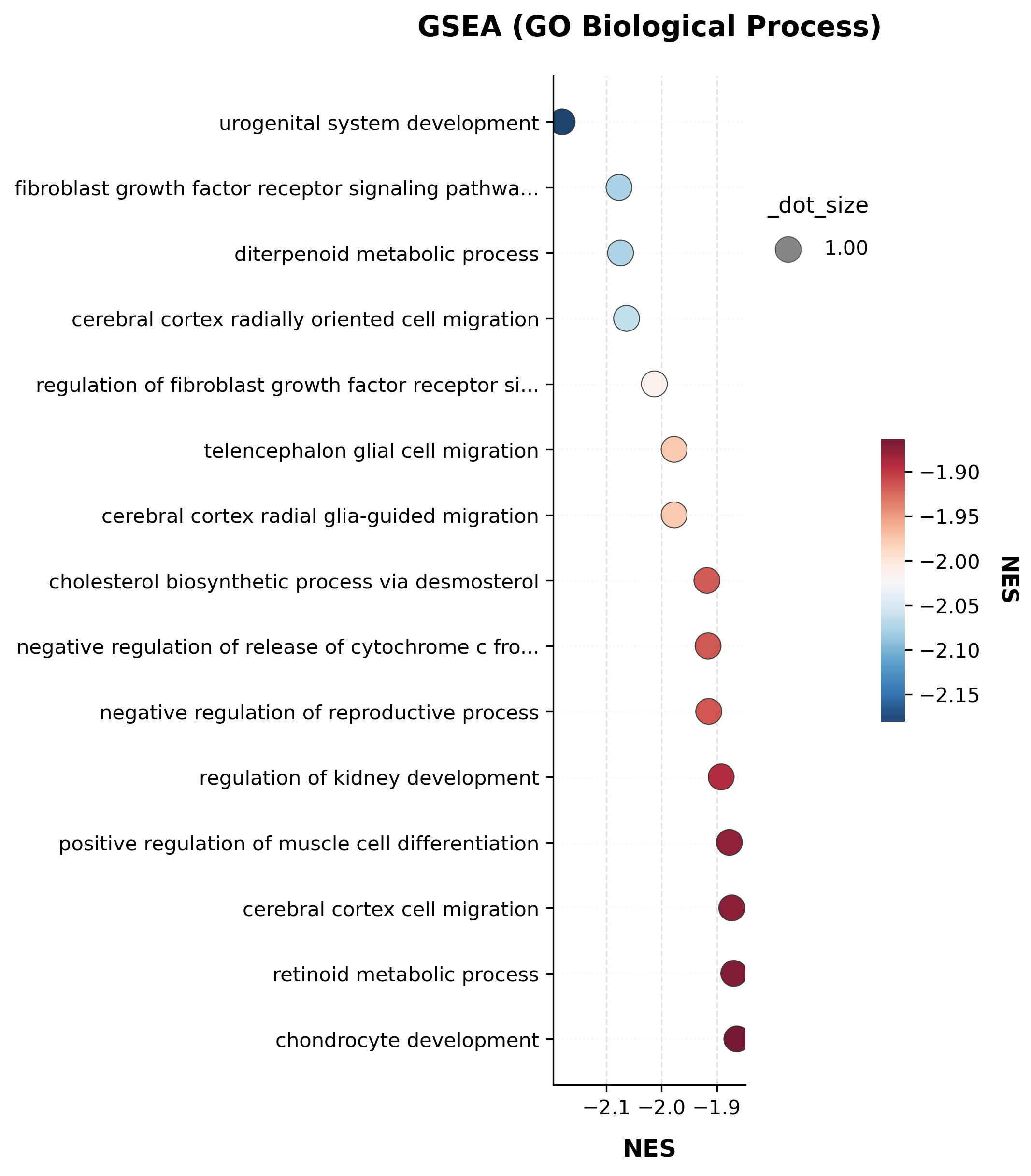
gsea_res.sort_values("p.adjust")[["Term", "NES", "pvalue", "p.adjust", "leading_edge"]].head(10)
2026-07-05 11:40:29,063 [WARNING] Duplicated values found in preranked stats: 87.82% of genes
The order of those genes will be arbitrary, which may produce unexpected results.
| Term | NES | pvalue | p.adjust | leading_edge | |
|---|---|---|---|---|---|
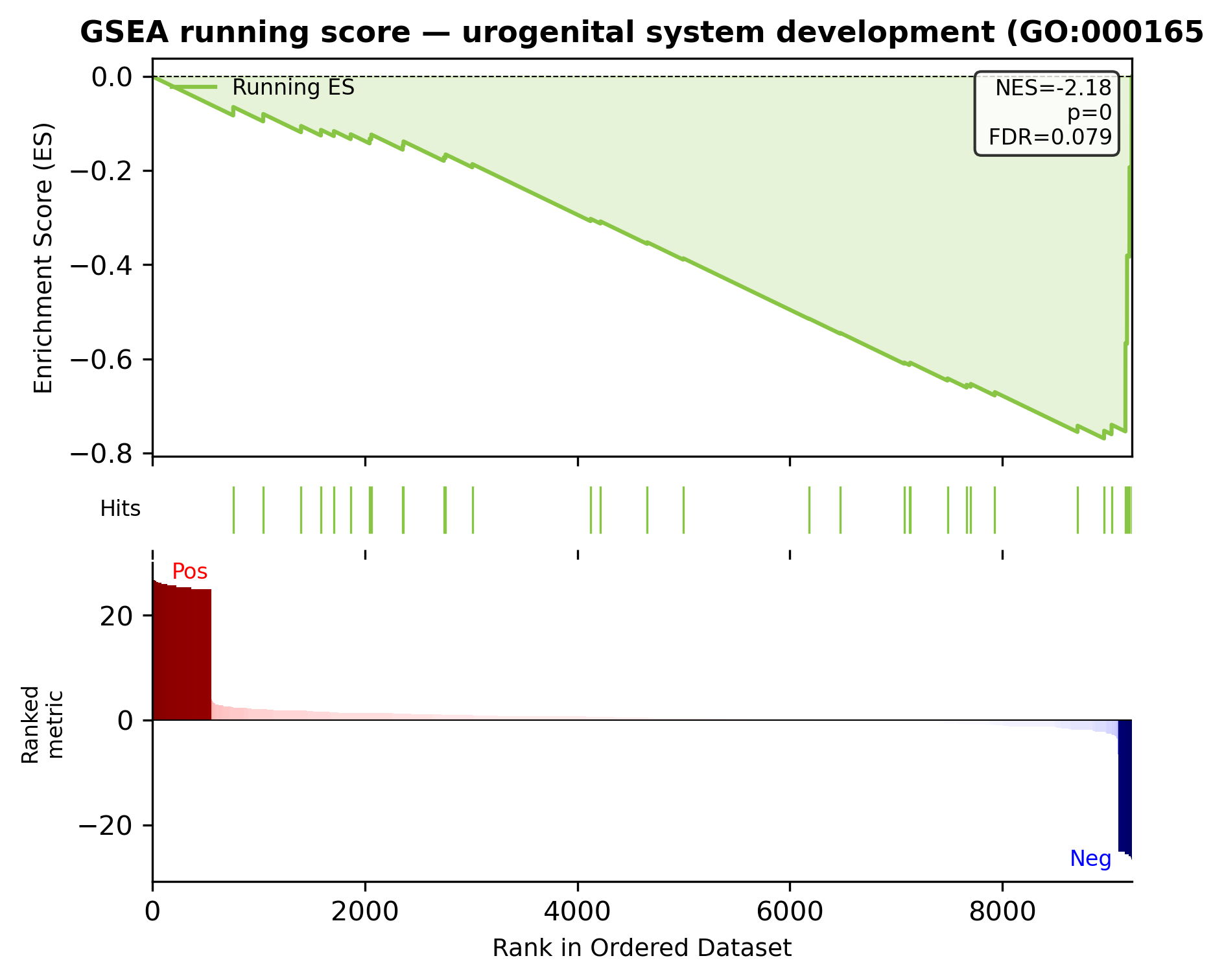
| 0 | urogenital system development (GO:0001655) | -2.180388 | 0.0 | 0.079007 | Igf1;Acd;Osr1;Sulf1;Serpinf1;Acvr1 |
| 3 | cerebral cortex radially oriented cell migrati... | -2.063498 | 0.0 | 0.102709 | Bmerb1;Lamb1;Dcx;Fbxo45;Reln;Col3a1 |
| 4 | regulation of fibroblast growth factor recepto... | -2.013076 | 0.0 | 0.116930 | Smoc2;Runx2;Sulf1;Wnt4 |
| 2 | diterpenoid metabolic process (GO:0016101) | -2.074478 | 0.0 | 0.121144 | Prmt3;Dgat1;Aldh1a1;Rbp4;Dhrs4;Cyp1b1 |
| 5 | telencephalon glial cell migration (GO:0022030) | -1.977310 | 0.0 | 0.137698 | Bmerb1;Lamb1;Dcx;Reln;Col3a1 |
| 6 | cerebral cortex radial glia-guided migration (... | -1.977310 | 0.0 | 0.137698 | Bmerb1;Lamb1;Dcx;Reln;Col3a1 |
| 1 | fibroblast growth factor receptor signaling pa... | -2.077310 | 0.0 | 0.181716 | Sos1;Smoc2;Runx2;Sulf1;Wnt4;Fgfr2;Fgf9;Ccn2;Fg... |
| 9 | negative regulation of reproductive process (G... | -1.914572 | 0.0 | 0.205418 | Igf1;Shb;Sulf1;Ptgds;Serpinf1;Wnt4 |
| 8 | negative regulation of release of cytochrome c... | -1.915757 | 0.0 | 0.228242 | Igf1 |
| 14 | chondrocyte development (GO:0002063) | -1.863570 | 0.0 | 0.243341 | Runx2;Sulf1 |
Note
Both scat.pl.gseaplot(...) (the running-sum plot) and scat.pl.enrich_dotplot(gsea_res, x="NES", ...) are shown below. An earlier draft of this tutorial found these calls effectively hung (>1 minute, no error) on a real, thousands-of-terms GSEA result. The root cause turned out to be a real scATrans performance bug, since fixed: run_gsea stored gseapy’s full per-term running-enrichment-score curves in gsea_res.attrs["gsea_details"], and pandas deep-copies .attrs on essentially every DataFrame operation (.head(), .copy(), slicing) via NDFrame.__finalize__ — for a genome-wide ranked list against thousands of gene sets that is tens of millions of floats being deep-copied on every such call. See CHANGELOG.md for the fix (the payload is now wrapped so its __deepcopy__ is an O(1) identity return).
scat.pl.enrich_dotplot(gsea_res, top_n=15, x="NES", color_by="NES", title="GSEA (GO Biological Process)");
top_term = gsea_res.iloc[0]["Term"]
scat.pl.gseaplot(ranked, gsea_res, term=top_term, title=f"GSEA running score — {top_term[:40]}");
simplified_jaccard = scat.simplify_enrichment(
go_res, method="jaccard", similarity_cutoff=0.5, min_count=3,
)
print(f"jaccard: {len(go_res)} -> {len(simplified_jaccard)} terms")
simplified_denester = scat.simplify_enrichment(
go_res, method="pathway_denester", min_count=3,
)
print(f"pathway_denester: {len(go_res)} -> {len(simplified_denester)} terms")
jaccard: 6744 -> 162 terms
pathway_denester: 6744 -> 130 terms
Comparing gene sets: compare_enrichment + concat_compare_results#
compare_enrichment runs ORA across several named gene lists at once
(clusterProfiler’s compareCluster style) and returns a single table with a
Cluster column — exactly the shape enrich_upsetplot / enrich_vennplot
expect. Here we compare the up- vs. down-regulated candidate genes.
cmp_res = scat.compare_enrichment(
{"up": candidate_genes, "down": down_genes},
organism="mouse",
gene_sets="GO_Biological_Process",
adata=adata_norm,
)
cmp_res["Cluster"].value_counts()
Cluster
up 47
Name: count, dtype: int64
concat_compare_results does the same thing when you already have
separate result tables in hand (e.g. combining our GO and KEGG results
above into one comparable table):
concat_res = scat.concat_compare_results({"GO": go_res, "KEGG": kegg_res})
concat_res["Cluster"].value_counts()
Cluster
GO 6744
KEGG 202
Name: count, dtype: int64
Exporting results#
save_enrichment_report writes CSV/TSV/Excel + a metadata JSON (gene-set
provenance, universe info, package version); expand_enrichment_genes
pivots to one row per gene for network/follow-up analyses. We write to a
scratch temp directory here rather than into the docs tree.
import tempfile, os
tmpdir = tempfile.mkdtemp()
saved_paths = scat.save_enrichment_report(
kegg_res,
prefix=os.path.join(tmpdir, "kegg_demo"),
save_excel=True, save_csv=True, save_tsv=True,
save_metadata=True, save_term_gene_table=True,
)
saved_paths
{'results_csv': '/tmp/tmplqv4ijao/kegg_demo_results.csv',
'term_gene_table_csv': '/tmp/tmplqv4ijao/kegg_demo_term_gene_table.csv',
'results_tsv': '/tmp/tmplqv4ijao/kegg_demo_results.tsv',
'term_gene_table_tsv': '/tmp/tmplqv4ijao/kegg_demo_term_gene_table.tsv',
'metadata_json': '/tmp/tmplqv4ijao/kegg_demo_metadata.json',
'results_xlsx': '/tmp/tmplqv4ijao/kegg_demo_results.xlsx'}
long_table = scat.expand_enrichment_genes(kegg_res)
print(long_table.shape)
long_table.head()
(146, 14)
| Term | Description | Gene | Count | GeneRatio | GeneRatio_str | BgRatio | BgRatio_str | FoldEnrichment | RichFactor | Overlap | pvalue | p.adjust | TermSize | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | Ribosome(mmu03010) | Rpl14 | 21 | 0.333333 | 21/63 | 0.029197 | 84/2877 | 11.416667 | 0.25 | 21/84 | 4.654655e-18 | 9.402402e-16 | 84 | |
| 1 | Ribosome(mmu03010) | Rpl18a | 21 | 0.333333 | 21/63 | 0.029197 | 84/2877 | 11.416667 | 0.25 | 21/84 | 4.654655e-18 | 9.402402e-16 | 84 | |
| 2 | Ribosome(mmu03010) | Rpl21 | 21 | 0.333333 | 21/63 | 0.029197 | 84/2877 | 11.416667 | 0.25 | 21/84 | 4.654655e-18 | 9.402402e-16 | 84 | |
| 3 | Ribosome(mmu03010) | Rpl23a | 21 | 0.333333 | 21/63 | 0.029197 | 84/2877 | 11.416667 | 0.25 | 21/84 | 4.654655e-18 | 9.402402e-16 | 84 | |
| 4 | Ribosome(mmu03010) | Rpl26 | 21 | 0.333333 | 21/63 | 0.029197 | 84/2877 | 11.416667 | 0.25 | 21/84 | 4.654655e-18 | 9.402402e-16 | 84 |
Plotting gallery#
Every scat.pl.* function below accepts ax=, save_path=, show=,
figsize=, and use_style= for embedding in multi-panel publication
figures (see Visualization).
Volcano plots (3 styles)#
scat.pl.volcano_plot(de_wilcoxon, style="auto", top_n=10, title="Volcano — auto (legacy) style");
scat.pl.volcano_plot(
de_wilcoxon, style="ggvolcano", top_n=10, logfc_cutoff=0.5, pval_cutoff=0.2,
title="Volcano — ggVolcano style",
);
scat.pl.volcano_plot(de_wilcoxon, style="gradual", top_n=10, title="Volcano — gradual (FDR gradient) style");
Enrichment dot plots and bar plot#
scat.pl.enrich_dotplot(go_res, top_n=15, title="GO Biological Process (ORA)");
scat.pl.enrich_dotplot(kegg_res, top_n=10, title="KEGG pathways (ORA)");
scat.pl.enrich_barplot(go_res, top_n=12, title="GO Biological Process (bar)");
UpSet and Venn: comparing up- vs. down-regulated enrichment#
Built for compare_enrichment/concat_compare_results output (a Cluster
column identifies each group).
scat.pl.enrich_upsetplot(cmp_res, pval_cutoff=0.05, title="Enriched GO terms: up vs. down candidates");
scat.pl.enrich_vennplot(cmp_res, pval_cutoff=0.05, title="Enriched GO terms: up vs. down candidates");

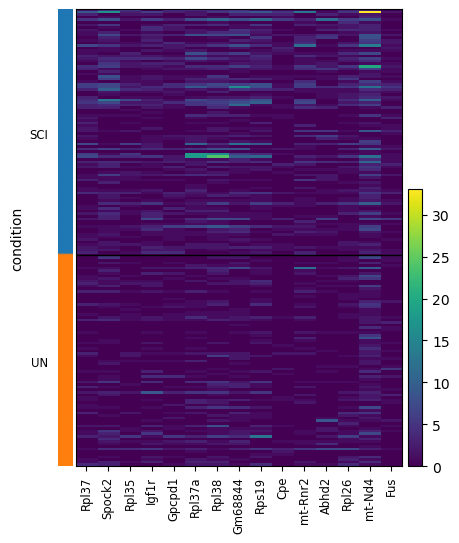
Heatmap of top candidate genes#
scat.pl.active_genes_heatmap(adata_norm, candidate_genes[:15], groupby="condition");
Not applicable here#
comet_plot, bias_diagnostic_plot, active_score_rankplot,
velocity_phase_portraits, and gamma_shrinkage_plot all require
active_score/unspliced_excess_*/effective_gamma columns that only
exist on the velocity-aware active_score path — see the sibling tutorial
Active Transcription Scoring on Real Spinal Cord Injury Data for those.
Recap#
Without ever touching a spliced/unspliced layer, this notebook covered:
Three DE backends (
de_method="wilcoxon",use_pseudobulk=True+ PyDESeq2,use_memento_de=True) on the same real SCI vs. UN comparison.Honest reporting of low per-gene DE power at n=3/group, with an exploratory-ranking workaround.
The full enrichment toolkit:
run_enrichment,run_kegg,run_go(ontology="ALL"),run_gsea,simplify_enrichment(both methods),compare_enrichment/concat_compare_results, and the export helpers.The plotting gallery: volcano (3 styles), enrichment dot/bar plots (including the GSEA NES dotplot and running-score plot), UpSet/Venn comparison plots, and a marker-gene heatmap.
See also: Standalone Differential Expression (no velocity data required), Functional Enrichment, Visualization, Statistical Guidance & Reporting Checklist, References & Data Sources, and Active Transcription Scoring on Real Spinal Cord Injury Data if your data does have spliced/unspliced layers.