Active Transcription Scoring on Real Spinal Cord Injury Data#
This tutorial runs the full active_score workflow — the velocity-aware
path for data with spliced/unspliced (or mature/nascent) layers — on a
real dataset: endothelial cells (EC) from adult mouse spinal cord, comparing
uninjured controls (UN, 3 replicates) against spinal cord injury (SCI,
3 replicates).
The data are a subset of:
Squair, J.W., Gautier, M., Kathe, C., Anderson, M.A., James, N.D., Hutson, T.H., Hudelle, R., Qaiser, T., Matson, K.J.E., Barraud, Q., Levine, A.J., La Manno, G., Skinnider, M.A., Courtine, G. (2021). Confronting false discoveries in single-cell differential expression. Nature Communications 12, 5692. DOI: 10.1038/s41467-021-25960-2 (GEO GSE165003). See References & Data Sources for the full citation.
Note
This choice of dataset is not an accident. Squair et al. (2021) is about
the false-discovery risk of treating single cells as independent replicates
in differential expression — exactly the pseudoreplication problem that
motivates scATrans’s use_pseudobulk, use_mixed_model, and
use_permutation machinery. We will see this play out directly on real
data below: a small (3 vs. 3 sample) design has genuinely limited power,
and the honest result is that few or no genes clear a strict FDR threshold.
We show that outcome as-is rather than manufacturing a cleaner-looking
example.
What active_score computes: for each gene, a composite score combining
(1) standard differential expression (logFC, adjusted p-value) with (2) a
reference-gamma unspliced excess term — how much more unspliced/nascent
RNA a gene shows in the target group than expected from a reference-group
U/S ratio — after optional bias correction for gene length and intron
count. The result is a ranking heuristic, not a p-value; see
Statistical Guidance & Reporting Checklist.
%matplotlib inline
import sys
sys.path.insert(0, "../../src") # use the in-repo scatrans, not any installed copy
import warnings
warnings.filterwarnings("ignore")
import numpy as np
import pandas as pd
import scanpy as sc
import scatrans as scat
print("scatrans", scat.__file__)
sc.settings.verbosity = 1
scatrans /home/lieber/scATrans-main/docs/tutorials/../../src/scatrans/__init__.py
Load the data#
EC.h5ad already contains spliced / unspliced layers (plus kb_python’s
mature / nascent / ambiguous, which scATrans does not need here since
spliced / unspliced are already correctly named).
adata = sc.read_h5ad("../../EC.h5ad")
adata
AnnData object with n_obs × n_vars = 177 × 26451
obs: 'n_genes_by_counts', 'log1p_n_genes_by_counts', 'total_counts', 'log1p_total_counts', 'pct_counts_in_top_50_genes', 'pct_counts_in_top_100_genes', 'pct_counts_in_top_200_genes', 'pct_counts_in_top_500_genes', 'condition', 'sample', 'GSM_ID', 'total_counts_mt', 'pct_counts_mt', 'n_genes', 'doublet_score', 'predicted_doublet'
var: 'gene_symbol', 'mt', 'n_cells_by_counts', 'mean_counts', 'pct_dropout_by_counts', 'total_counts', 'n_cells'
uns: 'sample_colors', 'scrublet'
layers: 'ambiguous', 'mature', 'nascent', 'spliced', 'unspliced'
adata.obs[["condition", "sample", "GSM_ID"]].value_counts().sort_index()
condition sample GSM_ID
SCI rep1 GSM5024314 35
rep2 GSM5024315 23
rep3 GSM5024316 37
UN rep1 GSM5024317 27
rep2 GSM5024318 38
rep3 GSM5024319 17
Name: count, dtype: int64
Six real biological samples: 3 uninjured (UN) and 3 spinal-cord-injury
(SCI) mice, GSM-labeled individually. Note that the sample labels
(rep1/rep2/rep3) are reused across conditions but refer to
different animals (different GSM_ID) — not a paired/blocked design.
This is exactly the case scATrans’s default (non-paired) replicate handling
is built for.
print("layers:", list(adata.layers.keys()))
print("X is raw counts:", np.allclose(adata.X.data, np.round(adata.X.data)), "max =", adata.X.max())
layers: ['ambiguous', 'mature', 'nascent', 'spliced', 'unspliced']
X is raw counts: True max = 488.0
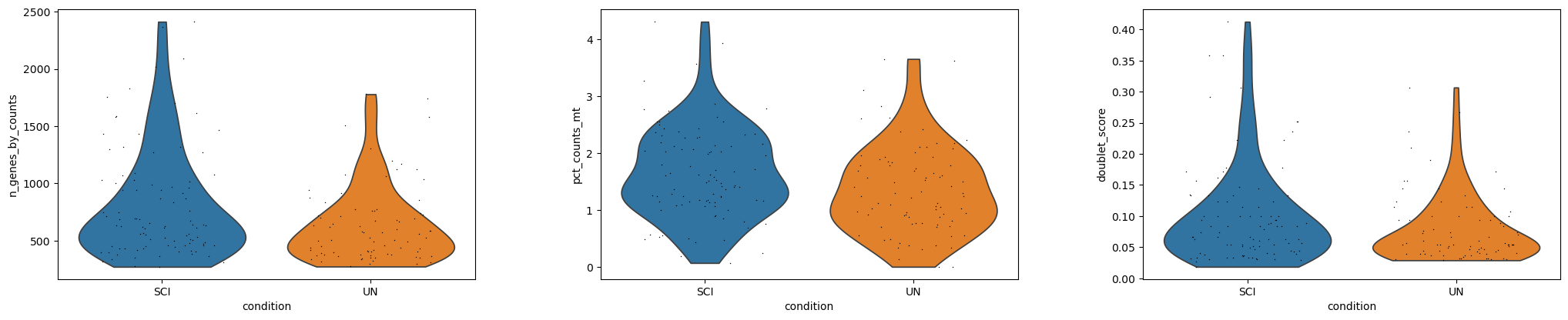
Quality control#
The object already carries QC metrics computed upstream (pct_counts_mt,
doublet_score, predicted_doublet, n_genes_by_counts). We inspect them
and apply light standard filters.
sc.pl.violin(
adata,
["n_genes_by_counts", "pct_counts_mt", "doublet_score"],
groupby="condition",
jitter=0.3,
multi_panel=True,
)
n_before = adata.n_obs
adata = adata[~adata.obs["predicted_doublet"]].copy()
adata = adata[adata.obs["pct_counts_mt"] < 20].copy()
adata = adata[adata.obs["n_genes_by_counts"] > 200].copy()
sc.pp.filter_genes(adata, min_cells=3)
print(f"cells: {n_before} -> {adata.n_obs}")
print(f"genes retained (>=3 cells): {adata.n_vars}")
cells: 177 -> 177
genes retained (>=3 cells): 9221
This particular dataset was already well curated upstream: none of the 177 cells are dropped by the doublet / mitochondrial / gene-count filters. The gene filter does matter, reducing the universe from 26,451 to the ~9.2k genes detected in at least 3 cells.
ufrac = scat.qc.unspliced_global(adata)
print(f"global unspliced fraction: {ufrac:.1%}")
global unspliced fraction: 30.2%
~30% unspliced is a healthy value (the package warns above ~50%, which
often signals a technical issue such as poor nuclear/cytoplasmic capture).
active_score re-runs and records this check automatically.
Preserve raw counts, then attach gene features#
store_raw_counts snapshots .X and the spliced/unspliced layers before
any normalization, so downstream count-based backends (PyDESeq2, Memento)
and enrichment background always have access to the full, unmodified gene
set. add_gene_features attaches the bundled mouse gene-length /
intron-count table used for optional bias correction.
scat.store_raw_counts(adata, layer="counts", save_raw=False)
adata = scat.add_gene_features(adata, organism="mouse")
adata.var[["gene_length", "intron_number"]].describe()
| gene_length | intron_number | |
|---|---|---|
| count | 9221.000000 | 9221.000000 |
| mean | 12915.754148 | 10.337599 |
| std | 15536.314470 | 9.969041 |
| min | 69.000000 | 0.000000 |
| 25% | 4750.000000 | 4.000000 |
| 50% | 9072.000000 | 8.000000 |
| 75% | 16191.000000 | 14.000000 |
| max | 321806.000000 | 145.000000 |
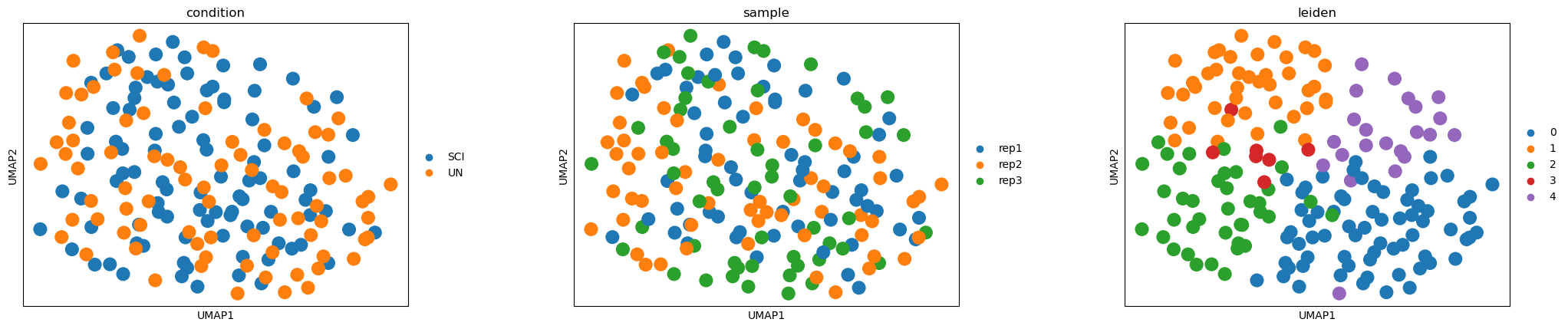
A quick look at cell states (optional, descriptive only)#
active_score itself works directly on .X plus the spliced/unspliced
layers — it does not require a UMAP or clustering. We compute one anyway,
purely to visualize how UN and SCI cells separate transcriptionally.
adata_vis = adata.copy()
sc.pp.normalize_total(adata_vis, target_sum=1e4)
sc.pp.log1p(adata_vis)
sc.pp.highly_variable_genes(adata_vis, n_top_genes=2000)
sc.pp.pca(adata_vis, use_highly_variable=True)
sc.pp.neighbors(adata_vis)
sc.tl.umap(adata_vis)
sc.tl.leiden(adata_vis, flavor="igraph", n_iterations=2)
sc.pl.umap(adata_vis, color=["condition", "sample", "leiden"], ncols=3, wspace=0.3)
diagnose_design: let the package tell you what design you have#
Before choosing options, run diagnose_design. It inspects cell counts,
sample counts, and the global unspliced fraction, and returns concrete
warnings and recommendations. It is also called automatically inside
active_score whenever sample_col or use_pseudobulk=True is supplied.
diag = scat.diagnose_design(
adata,
groupby="condition",
target_group="SCI",
reference_group="UN",
sample_col="sample",
)
print("n_cells (SCI / UN):", diag["n_cells_target"], "/", diag["n_cells_reference"])
print("n_samples (SCI / UN):", diag["n_samples_target"], "/", diag["n_samples_reference"])
print("suggested_preset:", diag["suggested_preset"])
print("workflow_preset:", diag["workflow_preset"])
print("\nwarnings:")
for w in diag["warnings"]:
print(" -", w)
print("\nrecommendations:")
for r in diag["recommendations"]:
print(" -", r)
n_cells (SCI / UN): 95 / 82
n_samples (SCI / UN): 3 / 3
suggested_preset: pseudobulk
workflow_preset: pseudobulk_report
warnings:
- Small number of biological samples per group (target=3, reference=3). Power for detecting differential nascent RNA excess will be limited. Permutation-based FDR is unreliable with so few label shuffles — prefer use_pseudobulk=True without permutation for ranking, then filter_active_genes (preset='pseudobulk') or DE p_adj for significance.
recommendations:
- With only 3 sample(s) per group, use use_pseudobulk=True (pseudobulk_de_backend='pydeseq2') rather than use_mixed_model=True (requires >=4 samples per group).
- After running active_score, always inspect adata.uns['scatrans']['diagnostics'] and the distributions in the returned all_results DataFrame before applying cutoffs.
With only 3 biological replicates per group, scATrans itself recommends pseudobulk + PyDESeq2 over a mixed model (which needs ≥4 samples/group) and flags that permutation-based FDR will be unreliable at this sample size. We follow that advice below, and also show what the single-cell heuristic and permutation paths look like for comparison.
Mode A — the one-liner: run_default_pipeline#
For a first look, run_default_pipeline scores, filters, and runs GO
enrichment in a single call, auto-selecting pseudobulk when enough
replicates are present.
result = scat.run_default_pipeline(
adata,
groupby="condition",
target_group="SCI",
reference_group="UN",
sample_col="sample",
organism="mouse",
)
print("auto-selected backend:", result["backend"])
print("filter preset used:", result["filter_preset"])
print("candidates:", len(result["candidates"]))
print("enrichment:", "None (no candidates passed the strict preset)" if result["enrichment"] is None else f"{len(result['enrichment'])} terms")
result["all_results"].sort_values("active_score", ascending=False).head()[["logFC", "p_adj", "active_score"]]
auto-selected backend: {'de_method': 't-test_overestim_var', 'use_pseudobulk': True, 'sample_col': 'sample', 'pseudobulk_de_backend': 'pydeseq2'}
filter preset used: pseudobulk
candidates: 0
enrichment: None (no candidates passed the strict preset)
| logFC | p_adj | active_score | |
|---|---|---|---|
| Vcam1 | 2.211801 | 0.999513 | 61.054032 |
| Cobll1 | 1.093613 | 0.999513 | 59.494934 |
| Tmeff2 | 1.194336 | 0.999513 | 58.849826 |
| Pitpnc1 | 1.018072 | 0.999513 | 58.295126 |
| Plcl1 | 0.962131 | 0.999513 | 57.720715 |
run_default_pipeline auto-selected pseudobulk + PyDESeq2 (because
sample_col was supplied with 3 replicates/group) and the matching
filter_preset="pseudobulk" — the same honest result as the manual walk-through
below: at p_adj < 0.05 on a 3-vs-3 design, zero genes pass, so
candidates is empty and enrichment is None (there is nothing to
enrich). This is expected, not an error — see Mode C for the
exploratory-candidate workaround and a full explanation.
Mode B — manual, single-cell heuristic#
active_score with its defaults runs differential expression per cell
(not per pseudobulk sample) and computes the unspliced-excess term directly
on single cells.
adata_res, significant, all_results = scat.active_score(
adata_input=adata,
groupby="condition",
target_group="SCI",
reference_group="UN",
de_method="wilcoxon",
show_plot=False,
)
all_results.sort_values("active_score", ascending=False).head(8)[
["logFC", "p_adj", "unspliced_excess_residual", "active_score"]
]
| logFC | p_adj | unspliced_excess_residual | active_score | |
|---|---|---|---|---|
| Prkg1 | 26.726971 | 0.994983 | 1.405985 | 66.727460 |
| Plcl1 | 27.793922 | 0.994983 | 0.782229 | 66.335979 |
| Samd12 | 26.600834 | 0.994983 | 0.602810 | 65.628986 |
| Nxn | 28.116873 | 0.994983 | 0.447306 | 64.068850 |
| Trpm3 | 2.930032 | 0.994983 | 1.073485 | 63.555366 |
| Gphn | 26.220245 | 0.994983 | 0.378836 | 62.809228 |
| Mecom | 2.117509 | 0.994983 | 1.124947 | 60.682844 |
| Cobll1 | 1.730982 | 0.994983 | 0.736788 | 58.014759 |
Note
Notice the extreme logFC values (~26-28) on a few genes (e.g. Prkg1,
Plcl1, Nxn). This is a well-known artifact of scanpy’s
rank_genes_groups log-fold-change estimator when a gene is expressed in
very few cells near zero in one group — not evidence of a huge biological
effect. active_score’s composite ranking is somewhat robust to this
because logFC is only one of three ingredients, but always sanity-check
individual genes against raw counts (e.g. with velocity_phase_portraits
below) before reporting them. See Statistical Guidance & Reporting Checklist.
print("heuristic-preset candidates:", len(scat.filter_active_genes(all_results, preset="heuristic")))
print("min p_adj across all genes:", all_results["p_adj"].min())
heuristic-preset candidates: 0
min p_adj across all genes: 0.9949828702539322
scat.pl.comet_plot(all_results, top_n=12, title="Active Drivers — single-cell heuristic (SCI vs UN)")
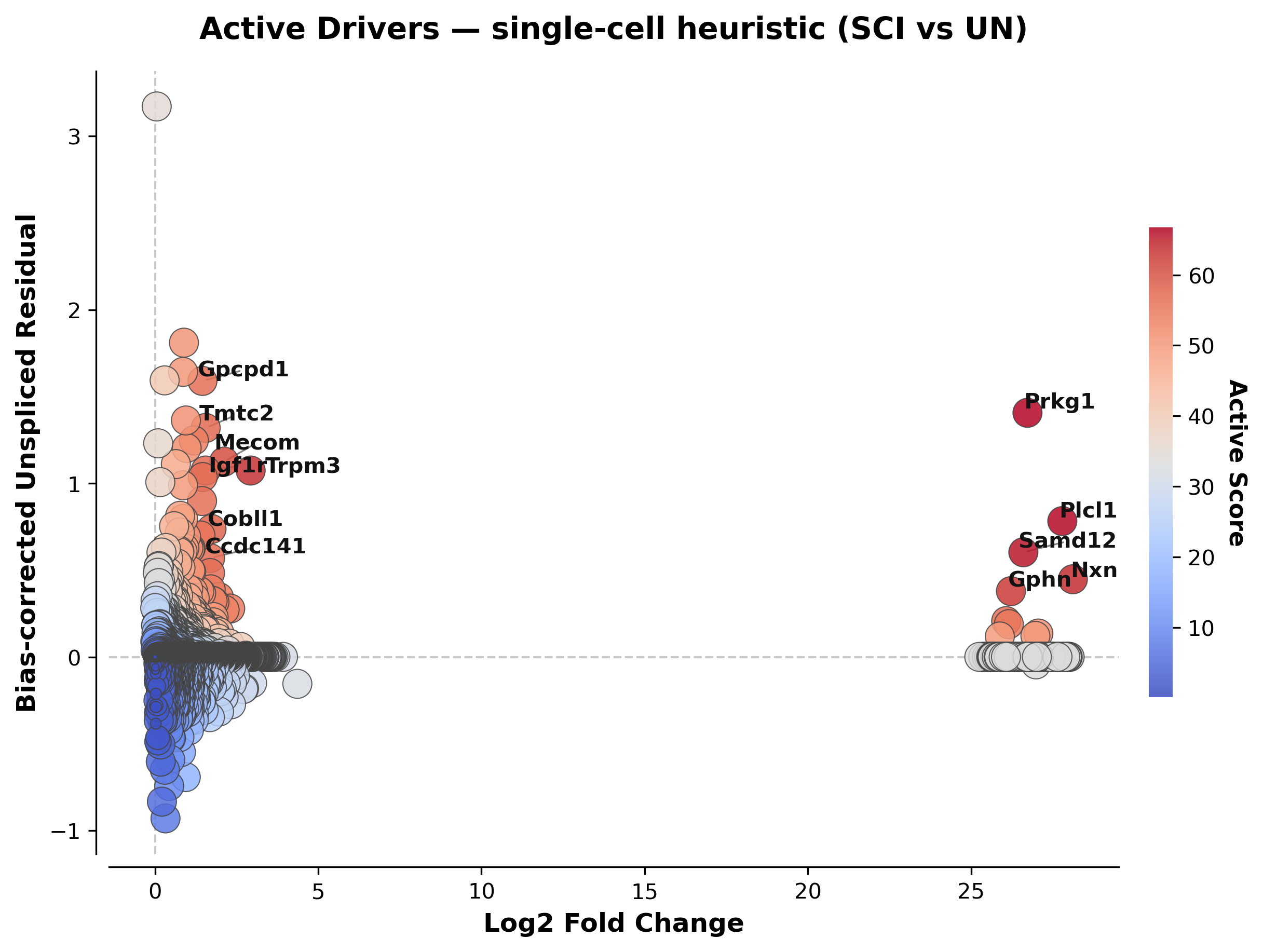
(<Figure size 2400x1800 with 2 Axes>,
<Axes: title={'center': 'Active Drivers — single-cell heuristic (SCI vs UN)'}, xlabel='Log2 Fold Change', ylabel='Bias-corrected Unspliced Residual'>)
Mode C — pseudobulk + PyDESeq2 (the recommended path here)#
This aggregates cells into one pseudobulk profile per (condition, sample)
and runs PyDESeq2 — the design diagnose_design recommended above.
adata_res_pb, significant_pb, all_results_pb = scat.active_score(
adata_input=adata,
groupby="condition",
target_group="SCI",
reference_group="UN",
use_pseudobulk=True,
sample_col="sample",
pseudobulk_de_backend="pydeseq2",
show_plot=False,
)
all_results_pb.sort_values("active_score", ascending=False).head(10)[
["logFC", "p_adj", "unspliced_excess_residual", "active_score"]
]
| logFC | p_adj | unspliced_excess_residual | active_score | |
|---|---|---|---|---|
| Vcam1 | 2.211801 | 0.999513 | 7.739810 | 61.054032 |
| Cobll1 | 1.093613 | 0.999513 | 23.042433 | 59.494934 |
| Tmeff2 | 1.194336 | 0.999513 | 11.158579 | 58.849826 |
| Pitpnc1 | 1.018072 | 0.999513 | 15.865926 | 58.295126 |
| Plcl1 | 0.962131 | 0.999513 | 16.528507 | 57.720715 |
| Igf1r | 0.899307 | 0.999513 | 31.005419 | 57.308573 |
| Zfp366 | 1.159273 | 0.999513 | 8.128796 | 56.450931 |
| Mecom | 0.866779 | 0.999513 | 14.552282 | 56.212011 |
| Tjp1 | 0.807648 | 0.999513 | 16.418442 | 55.620784 |
| Rapgef5 | 0.989955 | 0.999513 | 9.047048 | 55.518645 |
print("min p_adj (pseudobulk DESeq2):", all_results_pb["p_adj"].min())
print("preset='pseudobulk' candidates:", len(scat.filter_active_genes(all_results_pb, preset="pseudobulk")))
print("built-in `significant` list:", len(significant_pb))
min p_adj (pseudobulk DESeq2): 0.5584481513291821
preset='pseudobulk' candidates: 0
built-in `significant` list: 0
Note
With only 3 vs. 3 pseudobulk samples, no gene clears p_adj < 0.05
here (minimum adjusted p-value ≈ 0.56) — the built-in significant list
and the strict preset="pseudobulk" filter are both legitimately empty.
This is real, honest low-power biology, not a bug: three replicates per
group is thin for genome-wide multiple testing correction. The right move
is not to lower the threshold until something turns up, but to treat
active_score as a ranking/hypothesis-generating tool and say so plainly
in any write-up — see Statistical Guidance & Reporting Checklist.
# Exploratory ranking: effect size + active_score, p-value gate relaxed
# (this is a *ranked candidate list* for hypothesis generation, not a
# p<0.05 confirmatory claim — see the note above).
candidates = scat.filter_active_genes(
all_results_pb,
active_score_cutoff=40,
logfc_cutoff=0.5,
pval_cutoff=1.0,
)
print("exploratory candidates:", len(candidates))
candidates.sort_values("active_score", ascending=False).head(10)[
["logFC", "p_adj", "unspliced_excess_residual", "active_score"]
]
exploratory candidates: 64
| logFC | p_adj | unspliced_excess_residual | active_score | |
|---|---|---|---|---|
| Vcam1 | 2.211801 | 0.999513 | 7.739810 | 61.054032 |
| Cobll1 | 1.093613 | 0.999513 | 23.042433 | 59.494934 |
| Tmeff2 | 1.194336 | 0.999513 | 11.158579 | 58.849826 |
| Pitpnc1 | 1.018072 | 0.999513 | 15.865926 | 58.295126 |
| Plcl1 | 0.962131 | 0.999513 | 16.528507 | 57.720715 |
| Igf1r | 0.899307 | 0.999513 | 31.005419 | 57.308573 |
| Zfp366 | 1.159273 | 0.999513 | 8.128796 | 56.450931 |
| Mecom | 0.866779 | 0.999513 | 14.552282 | 56.212011 |
| Tjp1 | 0.807648 | 0.999513 | 16.418442 | 55.620784 |
| Rapgef5 | 0.989955 | 0.999513 | 9.047048 | 55.518645 |
scat.pl.volcano_plot(
all_results_pb,
style="ggvolcano",
top_n=12,
logfc_cutoff=0.5,
pval_cutoff=0.999, # relaxed to match the exploratory cutoff above (p_adj min here is ~0.56)
title="Pseudobulk PyDESeq2 — SCI vs UN",
)
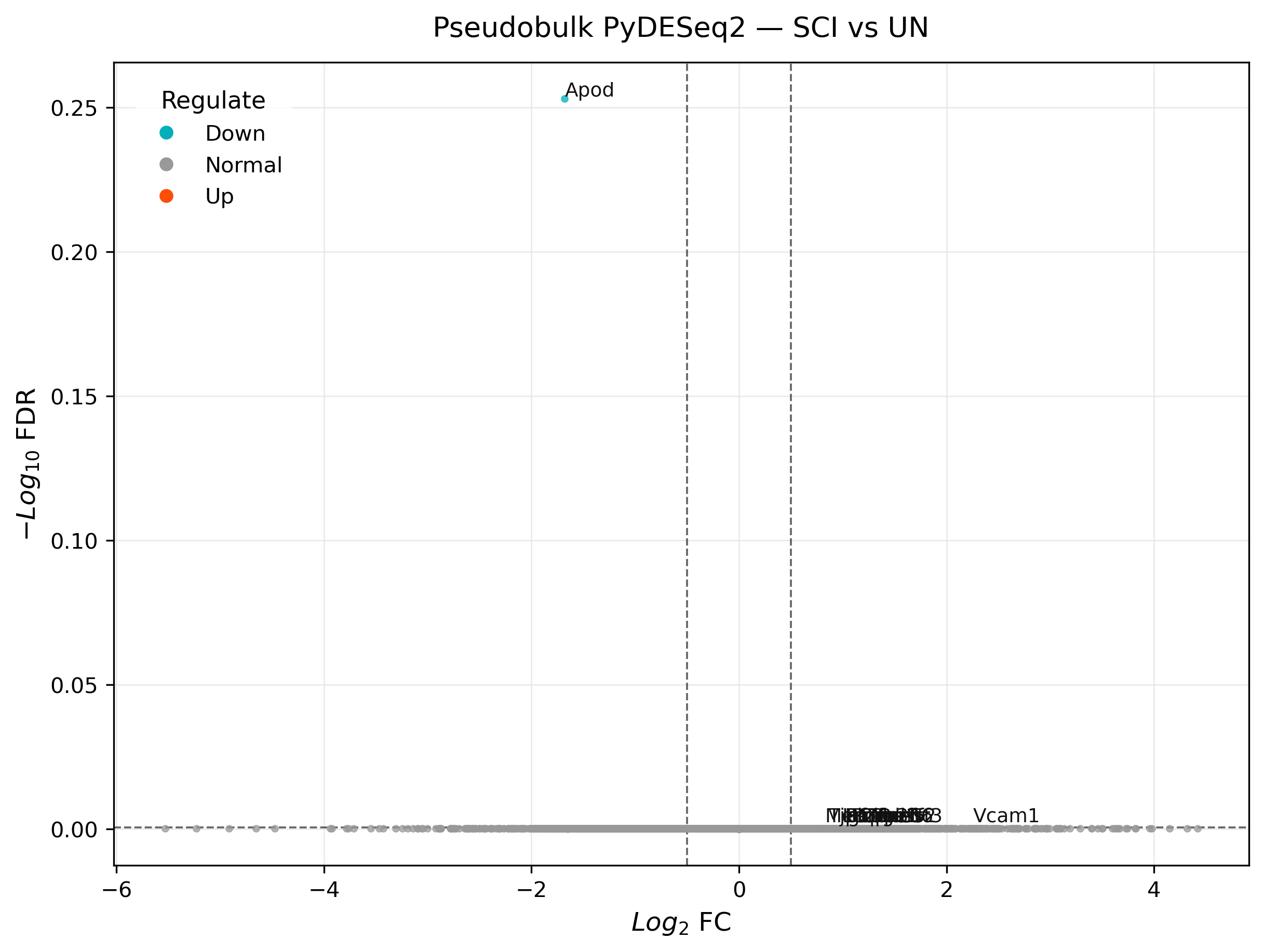
(<Figure size 2400x1800 with 1 Axes>,
<Axes: title={'center': 'Pseudobulk PyDESeq2 — SCI vs UN'}, xlabel='$Log_2$ FC', ylabel='$-Log_{10}$ FDR'>)
Mode D — permutation-based FDR (single-cell path)#
diagnose_design already warned that permutation is unreliable with only
3 samples/group in pseudobulk mode. We instead demonstrate the mechanism
on the single-cell heuristic path with a modest n_perm and the fast
backend, purely to show how it works — the conclusion is consistent with
everything above: still no significant genes at this sample size.
adata_res_perm, significant_perm, all_results_perm = scat.active_score(
adata_input=adata,
groupby="condition",
target_group="SCI",
reference_group="UN",
use_permutation=True,
n_perm=100,
perm_de_backend="fast", # exploratory speed; use "same" for manuscript-quality FDR
show_plot=False,
)
print("significant (permutation FDR):", len(significant_perm))
print(adata_res_perm.uns["scatrans"].get("permutation_approximation_note"))
all_results_perm.sort_values("active_score", ascending=False).head(5)[
["logFC", "p_adj", "unspliced_excess_fdr", "active_score"]
]
significant (permutation FDR): 0
For efficiency, unspliced/spliced layers and reference gamma are fixed from the original data. Group labels are shuffled to recompute DE, unspliced excess residual, composite active_score, and unspliced_excess permutation p-values.
| logFC | p_adj | unspliced_excess_fdr | active_score | |
|---|---|---|---|---|
| Plcl1 | 27.793922 | 0.498326 | 0.999048 | 74.963961 |
| Nxn | 28.116873 | 0.456581 | 1.000000 | 73.615214 |
| Prkg1 | 26.726971 | 0.587123 | 0.706809 | 73.537238 |
| Igf1r | 1.549713 | 0.212572 | 0.501937 | 73.407025 |
| Mecom | 2.117509 | 0.347586 | 0.632578 | 72.878279 |
Mode E — reference-gamma robustness and bias-correction diagnostics#
gamma_method="empirical_bayes" shrinks each gene’s reference U/S ratio
toward a shared, hierarchically-estimated prior — a more robust option when
the reference group is small. show_effective_gamma=True exposes the
per-gene shrunk gamma so its behavior can be inspected directly.
adata_res_gb, _, all_results_gb = scat.active_score(
adata_input=adata,
groupby="condition",
target_group="SCI",
reference_group="UN",
gamma_method="empirical_bayes",
show_effective_gamma=True,
show_plot=False,
)
v = adata_res_gb.uns["scatrans"]["diagnostics"]["velocity"]
print("gamma_prior_mean:", v.get("gamma_prior_mean"))
print("shrinkage_summary:", v.get("shrinkage_summary"))
gamma_prior_mean: -2.1783053057222053
shrinkage_summary: {'mean': 0.9630662745530777, 'q10': 0.94207253767107, 'q25': 0.9508108342897064, 'q50': 0.9590140129606987, 'q75': 0.9786133392259676, 'q90': 0.9873976599591713}
scat.pl.gamma_shrinkage_plot(all_results_gb)
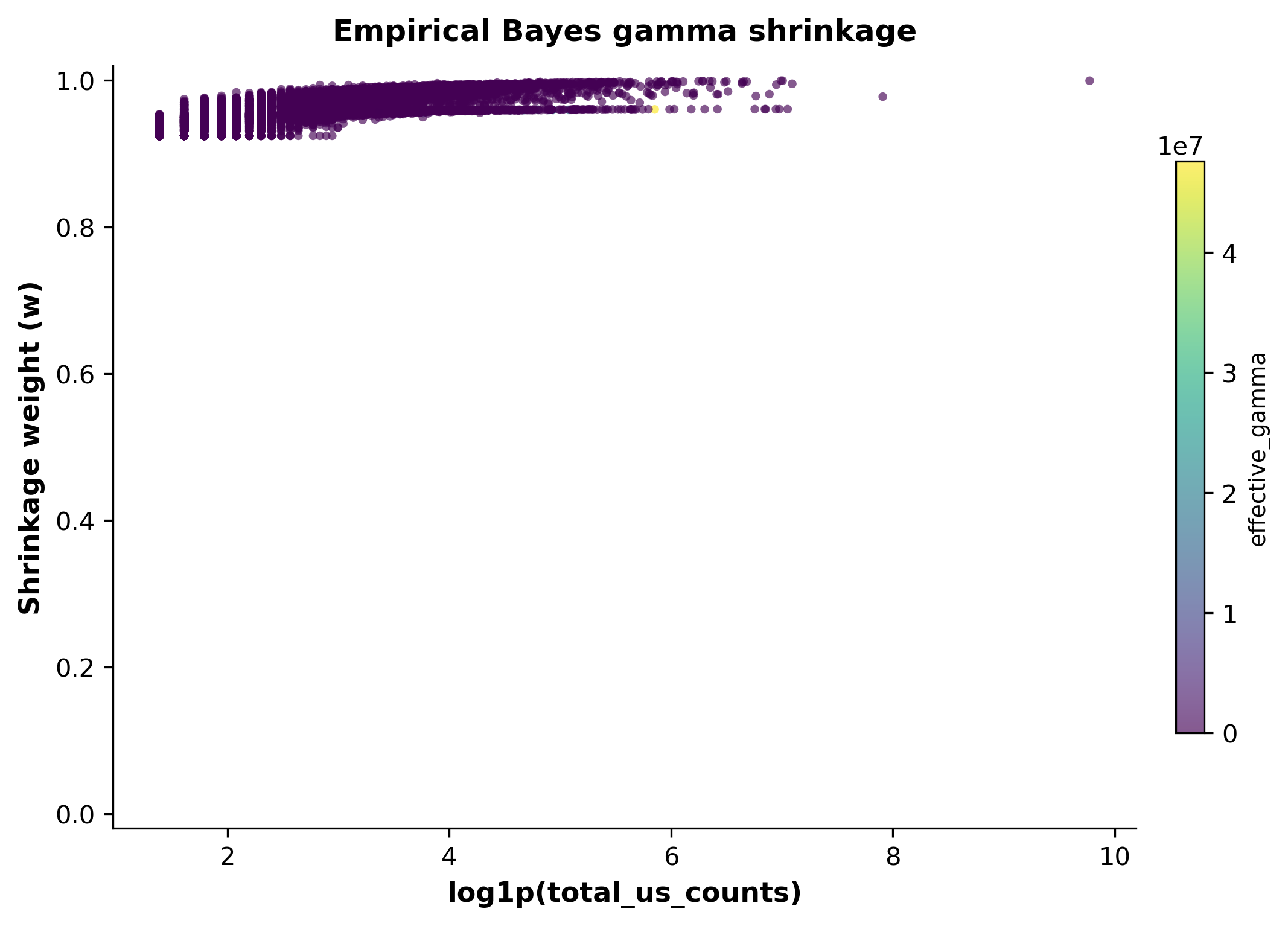
(<Figure size 2100x1500 with 2 Axes>,
<Axes: title={'center': 'Empirical Bayes gamma shrinkage'}, xlabel='log1p(total_us_counts)', ylabel='Shrinkage weight (w)'>)
scat.pl.bias_diagnostic_plot(all_results_gb, title="Length/intron bias correction — before vs. after")
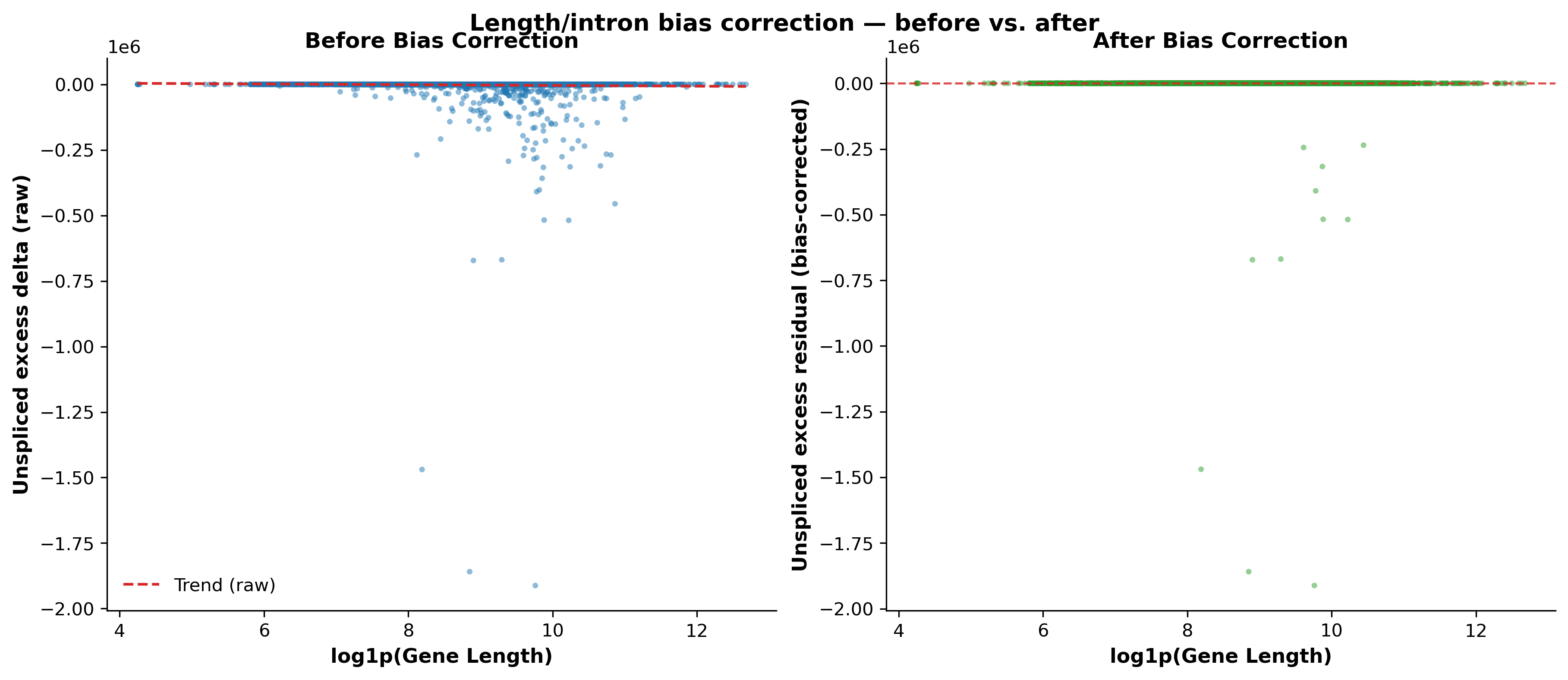
(<Figure size 3600x1500 with 2 Axes>,
array([<Axes: title={'center': 'Before Bias Correction'}, xlabel='log1p(Gene Length)', ylabel='Unspliced excess delta (raw)'>,
<Axes: title={'center': 'After Bias Correction'}, xlabel='log1p(Gene Length)', ylabel='Unspliced excess residual (bias-corrected)'>],
dtype=object))
Mode F — advanced mode (scVelo moments smoothing)#
mode="advanced" uses scVelo’s moments (kNN-smoothed spliced/unspliced
means) before computing the group-wise gamma delta, instead of the raw
per-cell counts. It is still a reference-gamma excess calculation, not a
full dynamical velocity model — just computed on smoothed rather than raw
values. Requires pip install "scatrans[advanced]".
adata_res_adv, _, all_results_adv = scat.active_score(
adata_input=adata,
groupby="condition",
target_group="SCI",
reference_group="UN",
mode="advanced",
show_plot=False,
)
all_results_adv.sort_values("active_score", ascending=False).head(5)[
["logFC", "p_adj", "active_score"]
]
computing moments based on connectivities
finished (0:00:00) --> added
'Ms' and 'Mu', moments of un/spliced abundances (adata.layers)
| logFC | p_adj | active_score | |
|---|---|---|---|
| Plcl1 | 27.793922 | 0.498326 | 74.586539 |
| Prkg1 | 26.726971 | 0.587123 | 73.507546 |
| Nxn | 28.116873 | 0.456581 | 73.222538 |
| Mecom | 2.117509 | 0.347586 | 72.813430 |
| Igf1r | 1.549713 | 0.212572 | 72.749187 |
Closing the loop: one enrichment pass#
Functional enrichment is covered in full in the companion tutorial, scATrans without spliced/unspliced layers: differential expression + enrichment + plotting (ORA, KEGG, GO all-ontology, GSEA, redundancy reduction, and the full plotting gallery). Here we just run one GO Biological Process over-representation test on the pseudobulk exploratory candidates from Mode C, to close the workflow end to end.
enrich_res = scat.run_enrichment(
gene_list=candidates.index.tolist(),
gene_sets="GO_Biological_Process",
organism="mouse",
adata=adata, # uses the full stored gene set (from store_raw_counts) as background
pval_cutoff=1.0, # relaxed, consistent with the exploratory candidate list above
)
print(enrich_res.shape)
enrich_res.sort_values("pvalue").head(10)[["Term", "Overlap", "pvalue", "p.adjust"]]
(13, 15)
| Term | Overlap | pvalue | p.adjust | |
|---|---|---|---|---|
| 4 | adherens junction organization (GO:0034332) | 4/33 | 0.000094 | 0.629774 |
| 0 | regulation of synapse organization (GO:0050807) | 8/253 | 0.000505 | 0.629774 |
| 2 | protein localization to adherens junction (GO:... | 2/5 | 0.000530 | 0.629774 |
| 5 | cell junction assembly (GO:0034329) | 9/321 | 0.000531 | 0.629774 |
| 3 | regulation of cell junction assembly (GO:1901888) | 7/195 | 0.000548 | 0.629774 |
| 1 | regulation of synapse structure or activity (G... | 8/257 | 0.000560 | 0.629774 |
| 6 | regulation of hormone metabolic process (GO:00... | 3/25 | 0.000788 | 0.742972 |
| 8 | cellular response to amyloid-beta (GO:1904646) | 3/26 | 0.000886 | 0.742972 |
| 7 | response to amyloid-beta (GO:1904645) | 3/27 | 0.000992 | 0.742972 |
| 9 | bile acid and bile salt transport (GO:0015721) | 2/7 | 0.001103 | 0.743692 |
if len(enrich_res):
scat.pl.enrich_dotplot(enrich_res, top_n=10, title="GO Biological Process — pseudobulk exploratory candidates")
else:
print("No enrichment terms to plot.")
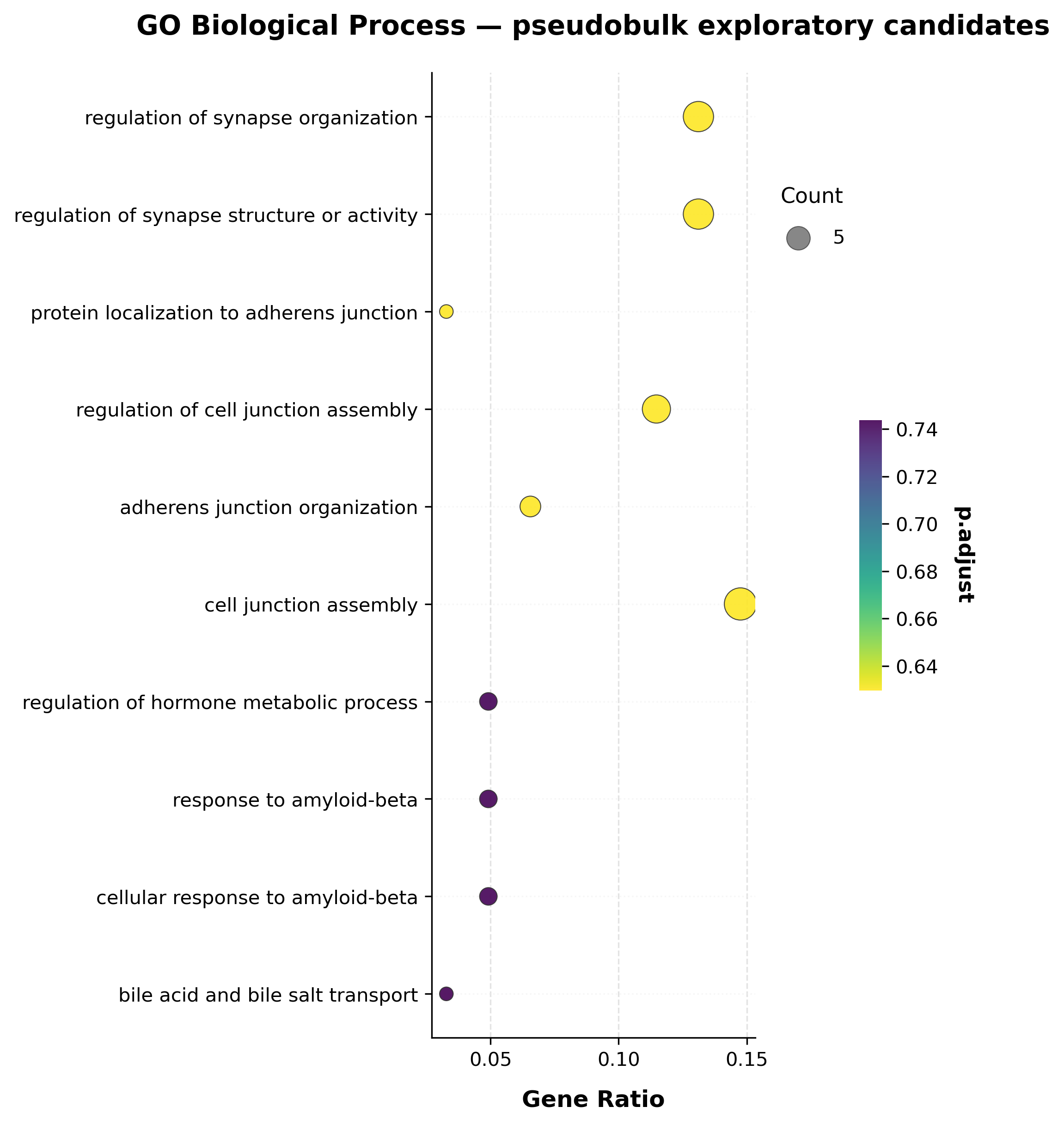
Rank plot and expression heatmap for the top candidates#
scat.pl.active_score_rankplot(all_results_pb.sort_values("active_score", ascending=False), top_n=15)
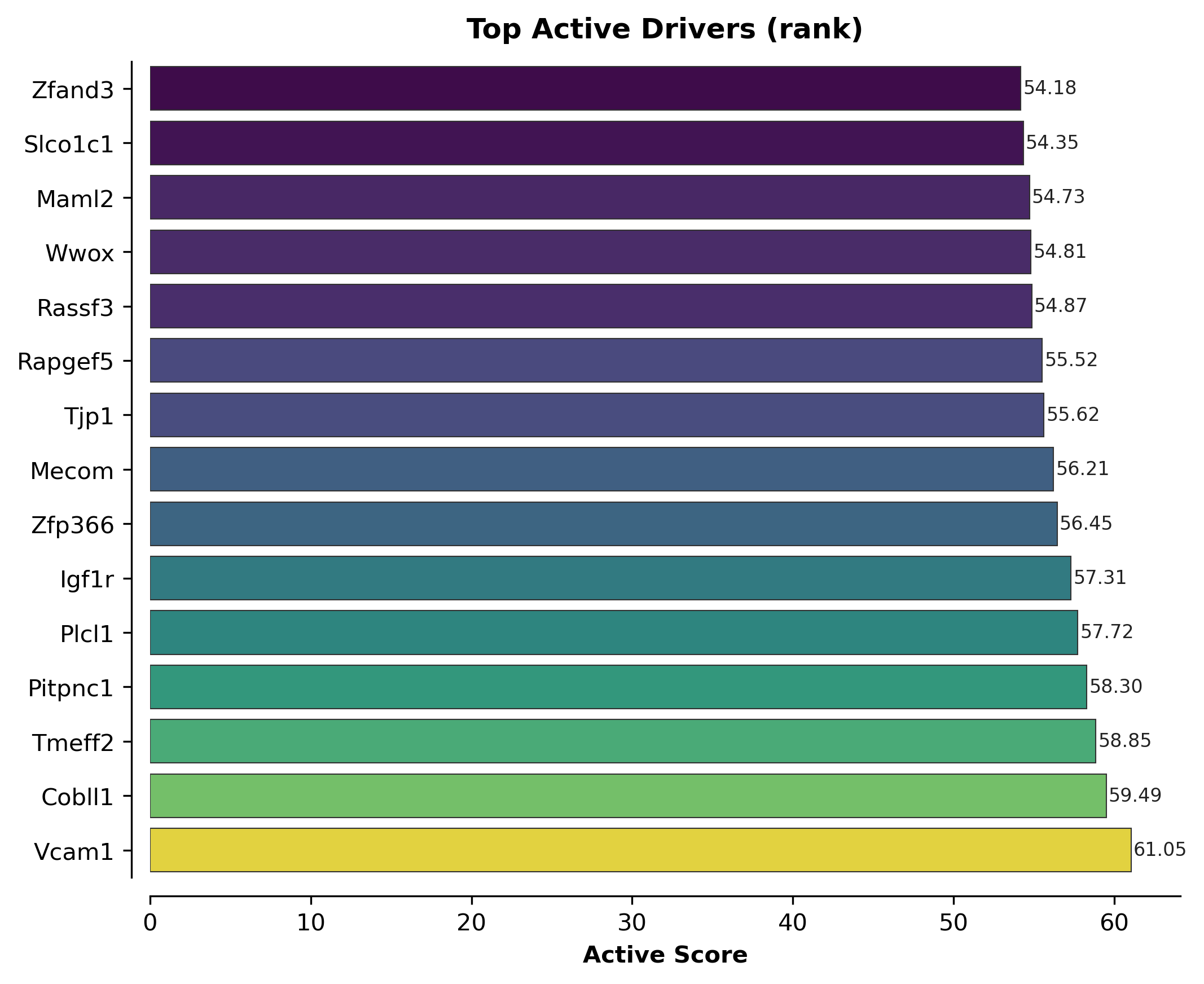
(<Figure size 2100x1710 with 1 Axes>,
<Axes: title={'center': 'Top Active Drivers (rank)'}, xlabel='Active Score'>)
top_genes = all_results_pb.sort_values("active_score", ascending=False).head(10).index.tolist()
scat.pl.active_genes_heatmap(adata_res_pb, genes=top_genes, groupby="condition")
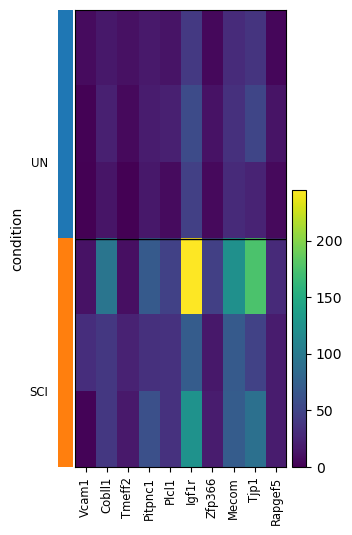
({'heatmap_ax': <Axes: >, 'groupby_ax': <Axes: ylabel='condition'>}, None)
scat.pl.velocity_phase_portraits(adata, genes=top_genes[:4], groupby="condition")
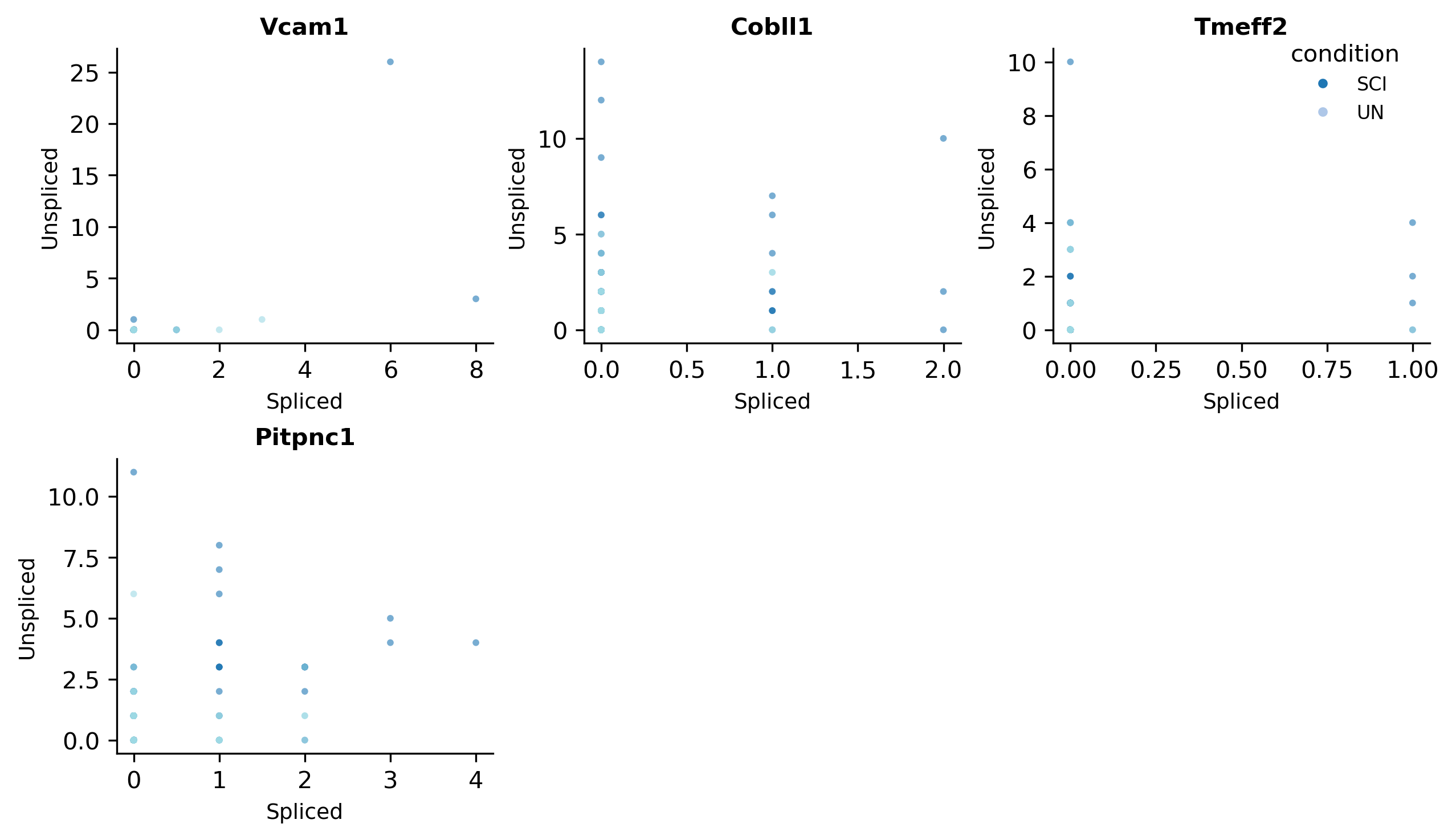
(<Figure size 2520x1440 with 6 Axes>,
array([<Axes: title={'center': 'Vcam1'}, xlabel='Spliced', ylabel='Unspliced'>,
<Axes: title={'center': 'Cobll1'}, xlabel='Spliced', ylabel='Unspliced'>,
<Axes: title={'center': 'Tmeff2'}, xlabel='Spliced', ylabel='Unspliced'>,
<Axes: title={'center': 'Pitpnc1'}, xlabel='Spliced', ylabel='Unspliced'>,
<Axes: >, <Axes: >], dtype=object))
Recap#
diagnose_design/recommend_workflowread the experimental design (here: 3 vs. 3 samples, ~30% unspliced) and steer you toward pseudobulk + PyDESeq2 rather than a mixed model or permutation FDR.On this real, modestly-powered dataset, strict significance filters (
preset="pseudobulk",preset="significant", permutation FDR) all legitimately return no genes — that is the honest answer for a 3 vs. 3 design, not a failure of the tool.active_scoreandfilter_active_geneswith relaxed, explicit cutoffs still produce a usable ranked candidate list for downstream enrichment and hypothesis generation.Always report
active_scoreas a composite heuristic rank, not a p-value — see Statistical Guidance & Reporting Checklist for the full reporting checklist, Optional Advanced Features for every option shown above, and References & Data Sources for the data and method citations.